
Заряд аккумулятора
Алгоритм заряда
Типы свинцово-кислотных аккумуляторов
На текущий момент на рынке аккумуляторов наиболее распространены следующие типы:
– SLA (Sealed Lead Acid) Герметичные свинцово-кислотные или VRLA (Valve Regulated Lead Acid) клапанно-регулируемые свинцово кислотные. Изготовлены по стандартной технологии. Благодаря конструкции и применяемых материалов, не требуют проверки уровня электролита и доливки воды. Имеют невысокую устойчивость к циклированию, ограниченные возможности работы при низком разряде, стандартный пусковой ток и быстрый разряд.
– EFB (Enhanced Flooded Battery) Технология разработана фирмой Bosch. Это промежуточная технология между стандартной и технологий AGM. От стандартной такие аккумуляторы отличаются более высокой устойчивостью к циклированию, улучшен прием заряда. Имеют более высокий пусковой ток. Как и у SLA\VRLA, есть ограничения работы при низкой заряженности.
– AGM (Absorbed Glass Mat) На текущий момент лучшая технология (по соотношению цена\характеристики). Устойчивость к циклированию выше в 3-4 раза, быстрый заряд. Благодаря низкому внутреннему сопротивлению обладает высоким пусковым током при низкой степени заряженности. Расход воды приближен к нулю, устойчива к расслоению электролита благодаря абсорбции в AGM-сепараторе.
– GEL (Gel Electrolite) Технология, при которой электролит находиться в виде геля. По сравнению с AGM обладают лучшей устойчивостью к циклированию, большая устойчивость к расслоению электролита. К недостаткам можно отнести высокую стоимость, и высокие требования к режиму заряда.
Существуют еще несколько технологий изготовления аккумуляторов, как связанных с изменением формы пластин, так и специфическими условиями эксплуатации.
Не смотря на различие технологий, физико-химические процессы протекающие при заряде — разряде аккумулятора одинаковые. По-этому алгоритмы заряда различных типов аккумуляторов практически идентичны.
Например, при заряде 12-ти вольтового аккумулятора по технологии:
– SLA\VRLA максимальный ток 0.1С, напряжение 14,2 … 14,5В
– AGM максимальный ток 0.2С, напряжение 14,6 … 14,8В
– GEL максимальный ток 0.2С, напряжение 14,1 … 14,4В
Значения приведены усредненные по рекомендациям различных производителей аккумуляторов. Конкретные значения необходимо уточнить у производителя.
Определение степени заряженности аккумулятора
Есть два основных способа определения степени заряженности аккумулятора, измерение плотности электролита и измерение напряжения разомкнутой цепи (НРЦ).
 0. При температуре аккумулятора 20-25оС (по рекомендации фирмы Bosch). Значения НРЦ приведены в таблице.
0. При температуре аккумулятора 20-25оС (по рекомендации фирмы Bosch). Значения НРЦ приведены в таблице.(у некоторых производителей значения могут отличаться от приведенных) Если степень заряженности аккумулятора меньше 80%, то рекомендуеться провести заряд.
Алгоритмы заряда аккумуляторов
Существуют несколько наиболее распространенных алгоритмов заряда аккумулятора. На текущий момент большинство производителей аккумуляторов рекомендуют алгоритм заряда CC\CV (Constant Current \ Constant Voltage – постоянный ток \ постоянное напряжение).
Такой алгоритм обеспечивает достаточно быстрый и «бережный» режим заряда аккумулятора. Для исключения долговременного пребывания аккумулятора в конце процесса заряда, большинство зарядных устройств переходит в режим поддержания (компенсации тока саморазряда) напряжения на аккумуляторе. Такой алгоритм называется трехступенчатым. График такого алгоритма заряда представлен на рисунке.
Указанные значения напряжения (14. 5В и 13.2В) справедливы при заряде аккумуляторов типа SLA\VRLA,AGM. При заряде аккумуляторов типа GEL значения напряжений должны быть установлены соответственно 14.1В и 13.2В.
5В и 13.2В) справедливы при заряде аккумуляторов типа SLA\VRLA,AGM. При заряде аккумуляторов типа GEL значения напряжений должны быть установлены соответственно 14.1В и 13.2В.
Дополнительные алгоритмы при заряде аккумуляторов
Предзаряд У сильно разряженного аккумулятора (НРЦ меньше 10В) увеличивается внутреннее сопротивление, что приводит к ухудшению его способности принимать заряд. Алгоритм предзаряда предназначен для «раскачки» таких аккумуляторов.
Асимметричный заряд Для уменьшения сульфатации пластин аккумулятора можно проводить заряд асимметричным током. При таком алгоритме заряд чередуется с разрядом, что приводит к частичному растворению сульфатов и восстановлению емкости аккумулятора.
Выравнивающий заряд В процессе эксплуатации аккумуляторов происходит изменение внутреннего сопротивления отдельных «банок», что в процессе заряда приводит неравномерности заряда. Для уменьшения разброса внутреннего сопротивления рекомендуется проводить выравнивающий заряд. При этом аккумулятор заряжают током 0.05…0.1C при напряжении 15.6…16.4В.
Заряд проводиться в течении 2…6 часов при постоянном контроле температуры аккумулятора. Нельзя проводить выравнивающий заряд герметичных аккумуляторов, особенно по технологии GEL. Некоторые производители допускают такой заряд для VRLA\AGM аккумуляторов.
При этом аккумулятор заряжают током 0.05…0.1C при напряжении 15.6…16.4В.
Заряд проводиться в течении 2…6 часов при постоянном контроле температуры аккумулятора. Нельзя проводить выравнивающий заряд герметичных аккумуляторов, особенно по технологии GEL. Некоторые производители допускают такой заряд для VRLA\AGM аккумуляторов.
Определение емкости аккумулятора
В процессе эксплуатации аккумулятора его емкость уменьшается. Если емкость составляет 80% от номинальной, то такой аккумулятор рекомендуется заменить. Для определения емкости аккумулятор полностью заряжают. Дают отстояться в течении 1….5 часов и затем разряжают током 1\20С до напряжения 10.8В (для 12-ти вольтового аккумулятора). Количество отданных аккумулятором ампер-часов является его фактической емкостью. Некоторые производители используют для определения емкости другие значения тока разряда, и напряжения до которого разряжается аккумулятор.
Контрольно-тренировочный цикл
Для уменьшения сульфатации пластин аккумулятора одна из методик это проведение контрольно тренировочных циклов (КТЦ). КТЦ состоят из нескольких последовательных циклов заряда с последующим разрядом током 0.01…0.05С. При проведении таких циклов, сульфат растворяется, емкость аккумулятора может быть частично восстановлена.
КТЦ состоят из нескольких последовательных циклов заряда с последующим разрядом током 0.01…0.05С. При проведении таких циклов, сульфат растворяется, емкость аккумулятора может быть частично восстановлена.
Схема зарядного устройства для восстановления АКБ реверсивным током
Всем привет, в этой статье поговорим о том, как собрать устройство для зарядки автомобильного аккумулятора реверсивным, ассиметричным током на полевых транзисторах.
Что такое зарядка АКБ реверсивным током, подробно останавливаться не буду, так как этой информации полно в инете. Для данного устройства было перепробовано много различных схем, большинство из них или не работало вообще, или работа остальных, тем или иным способом не устраивала по параметрам.
Поэтому пришлось начинать с нуля и сделать надёжную, работающую схему, что в конце концов и получилось. Вот так выглядит схема для зарядки аккумуляторов реверсивным током.Данная схема очень элементарна, очень надёжна и очень проста в повторении. Что мы видим на этой схеме, два 555-ых таймера включенных здесь в качестве генераторов импульсов. Каждая микросхема управляет своим полевым ключом.
Что мы видим на этой схеме, два 555-ых таймера включенных здесь в качестве генераторов импульсов. Каждая микросхема управляет своим полевым ключом.
Соответственно один мосфет отвечает за зарядку аккумулятора, второй мосфет за разрядку. Сначала давайте рассмотрим узел, который отвечает у нас за разрядку аккумулятора.555-ый таймер (№2) здесь настроен на частоту около 1Кгц с коэффициентом заполнения около 85%. Питание данной схемы осуществляется непосредственно от самого аккумулятора, именно поэтому в данной схеме очень важно использовать полевые транзисторы. Потому что в них присутствует, так называемый обратный диод, благодаря этому диоду и возможна работа данной схемы.
Вторая микросхема (№1) отвечает за зарядку аккумулятора, соответственно от того, как вы подберёте частота-задающую обвязку данной микросхемы и будет, в конечном итоге, зависеть время заряда и время разряда вашего аккумулятора.
Значит как же эта схема работает в целом…
Как только на выход нашего устройства мы подключаем какой-либо АКБ, соответственно у нас запускается микросхема №2 и начинает на своём выходе генерировать прямоугольные импульсы, в следствии чего у нас открывается транзистор VT2, который в свою очередь разряжает наш аккумулятор на какую-либо нагрузку, в моём случаи это автомобильная лампа на 21 ватт.
Микросхема под №1 у нас не запускается, так как на выходе нашего устройства стоит диод VD1 (сдвоенный диод-шоттки). На вход нашего устройства мы подключаем какой-либо источник питания, будь то зарядное устройство или какой-нибудь блок питания, соответственно у нас запускается микросхема под №1 и начинает также на своём выходе вырабатывать прямоугольные импульсы с той частотой с которой вы ей задали с помощью частота-задающей обвязки.И как только на выходе №1 микросхемы появляется высокий уровень у нас открываются транзисторы VT1 и VT3. Ну и как видно из схемы транзистор VT1 у нас закорачивает 5 вывод микросхемы №2 на землю, тем самым останавливая генерацию прямоугольных импульсов и запирая транзистор VT2, тем самым прекращая разрядку нашего аккумулятора.
И в то же время открытый транзистор VT3 соединяет наш аккумулятор с нашим источником питания, тем самым обеспечивая его зарядку.
Ну и соответственно, как только с выхода микросхемы №1 высокий уровень исчезает два транзистора VT1 и VT3 закрываются, тем самым разъединяя наше зарядное устройство от нашего аккумулятора и в то же время рассоединяя 5 вывод микросхемы №2 с землёй, тем самым восстанавливая генерацию прямоугольных импульсов на выходе.
По деталям…
Обе микросхемы питаются через 12-ти вольтовые стабилизаторы 7812.
Время заряда и время разряда АКБ можно регулировать изменяя номиналы резисторов R2,R3,R4 и частота-задающего конденсатора С3.
Плата получилась довольно компактная, мосфеты и диод установил на небольшой радиатор.
Хотя они работают в ключевом режиме и нагрев минимальный.
Клемники поставил для подключения разрядной лампы и аккумулятора.Вот подключил, загорелась лампочка, то есть пошла разрядка аккумулятора.Цикл разряда и цикл зарядаПоворачивая бегунок подстроечного резистора можно менять скорость заряда и разряда данной схемы.Данную платку можно разместить непосредственно в корпусе зарядного устройства, тем самым добавив ему очень полезную функцию десульфатации.
Печатку в формате .lay можно скачать здесь.
Зарядка Свинцово-Кислотных АКБ Асимметричным Током | PRACTICAL ELECTRONICS
Приветствую всех читателей канала! Сегодняшняя статья посвящена зарядному устройству для свинцово-кислотных аккумуляторов, которое использует асимметричный ток.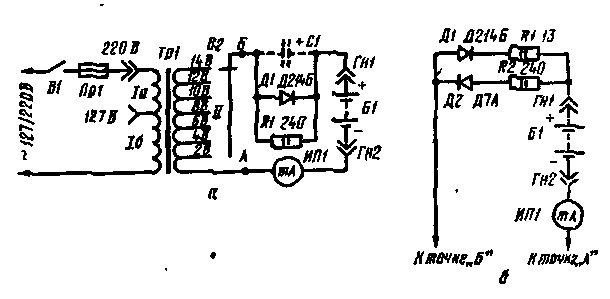 Ещё такое зарядное устройство известно под названием «реверсное». Тема эта довольно старая и есть разные схемные реализации данного процесса.
Ещё такое зарядное устройство известно под названием «реверсное». Тема эта довольно старая и есть разные схемные реализации данного процесса.
Но начнём с того, когда такое зарядное устройство целесообразно использовать, и в чём заключается принцип его действия. Зарядку асимметричным током используют в качестве профилактики для свинцово-кислотных АКБ от сульфатации или для уже частично сульфатированных аккумуляторов. Всё конечно зависит от степени «убитости» аккумулятора. А принцип довольно простой – на аккумулятор подаются положительные импульсы постоянного напряжения, которые его заряжают, а в перерывах их следования подключается нагрузка – мощный резистор или лампа накаливания. Асимметричный, или реверсивный, ток определяется наличием обратной амплитуды, иными словами, в каждом цикле он меняет своё направление. Количество электричества, протекающего при прямой полярности, больше, чем при обратной, что и обеспечивает заряд аккумулятора.
Примерный график заряда аккумулятора асимметричным (реверсным) током: Сз – емкость, сообщённая аккумулятору за время импульса tз; Ср – ёмкость, снятая с аккумулятора, за время импульса tp.
Примерный график заряда аккумулятора асимметричным (реверсным) током: Сз – емкость, сообщённая аккумулятору за время импульса tз; Ср – ёмкость, снятая с аккумулятора, за время импульса tp.
И ещё заряд аккумулятора реверсивным током дает возможность управлять восстановительными реакциями и структурными изменениями активного материала электродов. Меняя соотношения между зарядными и разрядными импульсами, а также изменяя их амплитуду, можно получать кристаллы сульфата свинца различных размеров и форм. Это позволяет увеличить пористость и суммарную площадь действующей поверхности электродов.
В некоторых подобных схемах в качестве коммутационного элемента, обеспечивающего переключение аккумулятора с заряда на разряд предлагается использовать электромагнитные реле. Но я не думаю, что это целесообразно с практической точки зрения, потому как контакты реле, особенно при значительных токах быстро придут в негодность. Практическая схема, где в качестве переключающих ключей используются транзисторы, показана на рисунке ниже. Это первый вариант схемы, которая предназначена для работы с внешним источником питания. В моём случае испытания проводились с лабораторным блоком питания, который ограничивал зарядный ток на уровне 5 А, а в качестве нагрузки использовалась двадцати-ваттная лампа накаливания 12 В.
Это первый вариант схемы, которая предназначена для работы с внешним источником питания. В моём случае испытания проводились с лабораторным блоком питания, который ограничивал зарядный ток на уровне 5 А, а в качестве нагрузки использовалась двадцати-ваттная лампа накаливания 12 В.
Схема электрическая принципиальная ЗУ для свинцово-кислотных АКБ асимметричным током
Схема работает от внешнего источника питания, которым обеспечивается зарядный ток для аккумулятора. Пока так. Хотелось бы узнать мнения радиолюбителей, кто занимался данным вопросом, особенно что касается длительности продолжительности зарядки и разрядки. А в дальнейшем схему можно доработать до полностью самостоятельного устройства с автоматическим отключением.
Генератор прямоугольных импульсов построен на базе таймера LM555 (NE555, КР1006ВИ1), который питается от стабилизатора напряжения DA1.
• среднее положение движка переменного резистора R2 – 40 сек и 10 сек;
• крайнее верхнее положение движка переменного резистора R2 – 30 сек и 30 сек;
• крайнее нижнее положение движка переменного резистора R2 – практически непрерывный высокий уровень с коротким (доли мс) низким уровнем.
Выход (3) таймера управляет двумя ключами на транзисторах VT1 и VT2. При высоком уровне напряжения отпирается VT1, и АКБ подключается к внешнему источнику питания. И наоборот, при низком уровне VT1 заперт, VT2 открыт и к АКБ подключена нагрузка (Rн).
Возможный вариант печатной платы показан на рисунке ниже. Транзисторы крепятся к теплоотводу, площадь которого следует выбирать исходя из величины зарядного/разрядного токов.
Для удобства навигации по разделу “Зарядные Устройства” подготовлена статья со ссылками на все опубликованные конструкции и кратким описанием. Ссылки будут добавляется по мере написания нового материала.
Восстановление автомобильного аккумулятора асимметричным током » Полезные самоделки
На рис. 1 приведено простое зарядное устройство, рассчитанное на использование вышеописанного способа. Схема обеспечивает импульсный зарядный ток до 10 А (используется для ускоренного заряда). Для восстановления и тренировки аккумуляторов лучше устанавливать импульсный зарядный ток 5 А. При этом ток разряда будет 0,5 А. Разрядный ток определяется величиной номинала резистора R4.
Рис. 1 Электрическая схема зарядного устройства.
Схема выполнена так, что заряд аккумулятора производится импульсами тока в течение одной половины периода сетевого напряжения, когда напряжение на выходе схемы превысит напряжение на аккумуляторе. В течение второго полупериода диоды VD1, VD2 закрыты и аккумулятор разряжается через нагрузочное сопротивление R4.
Значение зарядного тока устанавливается регулятором R2 по амперметру. Учитывая, что при зарядке батареи часть тока протекает и через резистор R4 (10%), то показания амперметра РА1 должны соответствовать 1,8 А (для импульсного зарядного тока 5 А), так как амперметр показывает усредненное значение тока за период времени, а заряд производится в течение половины периода.
В схеме предусмотрена защита аккумулятора от неконтролируемого разряда в случае случайного исчезновения сетевого напряжения. В этом случае реле К1 своими контактами разомкнет цепь подключения аккумулятора. Реле К1 применено типа РПУ-0 с рабочим напряжением обмотки 24 В или на меньшее напряжение, но при этом последовательно с обмоткой включается ограничительный резистор.
Для устройства можно использовать трансформатор мощностью не менее 150 Вт с напряжением во вторичной обмотке 22…25 В.
{banner_z}
Измерительный прибор РА1 подойдет со шкалой 0…5 А (0…3 А), например М42100. Транзистор VT1 устанавливаются на радиатор площадью не менее 200 кв. см, в качестве которого удобно использовать металлический корпус конструкции зарядного устройства.
Транзистор VT1 устанавливаются на радиатор площадью не менее 200 кв. см, в качестве которого удобно использовать металлический корпус конструкции зарядного устройства.
В схеме применяется транзистор с большим коэффициентом усиления (1000…18000), который можно заменить на КТ825 при изменении полярности включения диодов и стабилитрона, так как он другой проводимости. Последняя буква в обозначении транзистора может быть любой.
Рис. 2 Электрическая схема пускового устройства.
Для защиты схемы от случайного короткого замыкания на выходе установлен предохранитель FU2.
Резисторы применены такие R1 типа С2-23, R2 – ППБЕ-15, R3 – С5-16MB, R4 – ПЭВ-15, номинал R2 может быть от 3,3 до 15 кОм. Стабилитрон VD3 подойдет любой, с напряжением стабилизации от 7,5 до 12 В.
Приведенные схемы пускового (рис.2) и зарядного устройств (рис. 1) можно легко объединить (при этом не потребуется изолировать корпус транзистора VT1 от корпуса конструкции), для чего на пусковом трансформаторе достаточно намотать еще одну обмотку примерно 25. ..30 витков проводом ПЭВ-2 диаметром 1,8…2,0 мм.
Эта обмотка используется для питания схемы зарядного устройства.
Внимание!!! информация содержащаяся на данной странице, может быть устаревшей и содержать ошибки. Поэтому приводиться исключительно в ознакомительных целях.
Также есть быстрый способ восстановления автомобильного аккумулятора с помощью химических средств, Об этом читайте в этой статьеА
Зарядка аккумуляторов асимитричным током
Схема и описание самодельного зарядного устройства для зарядки автомобильных аккумуляторов асимметричным током.
Значительно лучших эксплуатационных характеристик аккумуляторов можно добиться, если их зарядку производить асимметричным током.
Схема устройства зарядки, реализующая такой принцип, показана на рисунке 1.
Рис.1. Нажмите на рисунок для просмотра.
При положительном полупериоде входного переменного напряжения ток протекает через элементы VD1, R1 и стабилизируется диодом VD2. Часть стабилизированного напряжения через переменный резистор R3 подается на базу транзистора VT2. Транзисторы VT2 и VT4 нижнего плеча устройства работают как генератор тока, величина которого зависит от сопротивления резистора R4 и напряжения на базе VT2. Зарядный ток в цепи аккумулятора протекает по элементам VD3, SA1.1, РА1, SA1.2, аккумулятор, коллекторный перепад транзистора VT4, R4.
При отрицательном полупериоде переменного напряжения на диоде VD1 работа устройства аналогична, но работает верхнее плечо – VD1 стабилизирует отрицательное напряжение, которое регулирует протекающий по аккумулятору ток в обратном напряжении (ток разрядки).
Показанный на схеме миллиамперметр РА1 используется при первоначальной настройке, в дальнейшем его можно отключить, переведя переключатель в другое положение.
Такое зарядное устройство обладает следующими преимуществами: 1. Зарядный и разрядный токи можно регулировать независимо друг от друга. Следовательно, в данном устройстве возможно применять аккумуляторы с различной величиной энергоемкости. 2. При каких-либо пропаданиях переменного напряжения каждое из плеч закрывается и через аккумулятор ток не протекает, что защищает аккумулятор от самопроизвольной разрядки.
В данном устройстве из отечественных элементов можно применить в качестве VD1 и VD2 – KC133A, VT1 и VT2 – КТ315Б или КТ503Б. Остальные элементы выбираются в зависимости от зарядного тока. Если он не превышает 100 мА, то в качестве транзисторов VT3 и VT4 следует применить КТ815 или КТ807 с любыми буквенными индексами (расположить на теплоотводе с площадью теплорассеивающей поверхности 5…15 кв.см), а в качестве диодов VD3 и VD4 – Д226, КД105 тоже с любыми буквенными индексами.
Читать далее – Как продлить работоспособность аккумуляторной батареи
Популярные схемы зарядных устройств:
Схема тиристорного зарядного устройства
Десульфатирующее зарядное устройство
Простое зарядное устройство
Схема автомата включения-выключения зарядного устройства
Ассиметричный электрохимический синтез веществ |НПК ЭНЕРГЕТИЧЕСКОЕ ОБОРУДОВАНИЕ
Ассиметричный электрохимический синтез веществ на переменном адаптивном токе в двойно электрическом слое.

Сильно засульфатированный аккумулятор не пригоден к работе. Мы восстанавливаем работоспособность засульфатированных электродов зарядом аккумуляторной батареи импульсным ассиметричным током.
Заряд импульсным ассиметричным током или методом импульсного заряда с обратным выбросом, характеризуется зарядом батареи импульсами с различной амплитудой с стабилизацией как по напряжению, так и по току между которыми есть короткие импульсы разрядного тока чередующимися rest-паузами, т.е ассиметричный ток – это переменный ток с различными амплитудами и длительностями импульсов обеих полярностей. За каждый период следования импульсов тока аккумулятор заряжается и частично разряжается. При определенном соотношении амплитуд и длительности импульсов прямого и обратного тока снижаются газовыделение и температура электролита.
График ассиметричного зарядаВ соответствии с теорией и практикой электрохимических процессов заряд аккумулятора ассиметричным током дает возможность управлять восстановительными реакциями и структурными изменениями активного материала электродов. Меняя соотношения между зарядными и разрядными импульсами, а также изменяя их амплитуду, можно получать кристаллы сульфата свинца различных размеров и форм. Это позволяет увеличить пористость и суммарную площадь действующей поверхности электродов, то есть увеличить поверхность соприкосновения электролита с активным материалом электродов, облегчить условия диффузии и выравнивания концентрации электролита в приэлектродном слое. Увеличение пористости способствует повышению величины максимального тока заряда и разряда.
Увеличение емкости при заряде аккумулятора асимметричным током происходит благодаря более эффективному окислению положительного электрода. При проведении импульсных режимов задается частота импульса. При увеличении частоты происходит изменение характера переднего и заднего фронтов импульса тока и соответственно напряжения аккумулятора. Величины амплитуд импульсов тока заряда и разряда определяются таким образом, чтобы получить оптимальное соотношение «время заряда/мощность зарядного устройства» и «время заряда/технико-эксплуатационные характеристики аккумулятора».
Оптимальной величиной соотношения импульсов заряда и разряда ( KI = I3 / I P ) является такая его величина, при которой напряжение на аккумуляторной батарее в течение паузы после импульса разряда остается постоянным. Оптимизация соотношения величин зарядного и разрядного импульсов приводит к снижению тепловыделения, а, следовательно, к возможности заряда большими токами и ускорению заряда.
Технология восстановления
Диагностика Оптимизация уровня электролита Регенерация Измерение остаточной емкости (разряд)Продление ресурса старой АКБУвелечение ресурса новой АКБСостояние положительных электродов щелочных никель-кадмиевых АКБ до восстановленияСостояние положительных электродов щелочных никель-кадмиевых АКБ после восстановленияВыравнивание напряжения на аккумуляторах Саморазряд АКБ снижен в 10 разПРЕИМУЩЕСТВА НАШЕЙ МЕТОДИКИ
Преимущества применения метода ассиметричного электрохимического синтеза веществ на переменном адаптивном токе в двойном электрическом слое очевидны:
- Экологически чистая технология.

- Без разборки аккумуляторов
- Без применения добавок и присадок.
- Энерго- и материало-сберегающая технология
- Экономия электроэнергии более чем в 2 раза по сравнению существующими зарядными устройствами.
- Эксплуатация восстановленных АКБ не отличается от требований изготовителей.
- Гарантия на восстановленные АКБ дается как на новые.
- Экономия денежных средств.
Десульфатация современных аккумуляторов.
Подавляющее большинство сегодняшних аккумуляторов необслуживаемые: не нужно следить за уровнем электролита, доливать воду. Однако, в процессе эксплуатации параметры автомобильных аккумуляторов ухудшаются: растет внутреннее сопротивление (уменьшается максимальный ток), снижается емкость. Свинцовые пластины по-прежнему разрушаются, сульфатируются.
Вероятно, Вы заметили, как
уменьшились размеры и вес современных автомобильных стартерных аккумуляторов? По
сравнению с аккумуляторами прошлого века? Как это достигается? Свинцовые
пластины стали намного тоньше, но их площадь увеличилась. Это привело к резкому
снижению прочности пластин.
Это привело к резкому
снижению прочности пластин.
Давно известен способ десульфатации аккумуляторов – зарядка асимметричным током. Давайте экспериментально проверим, как это работает. Современные схемы предполагают добавление импульсов повышенного напряжения. Например, известная схема. Возьмем её за основу, но увеличим мощность и добавим возможность менять параметры в широких пределах:
Катушка намотана на феррите от строчного трансформатора старого телевизора. 2 слоя ПЕЛ1,2мм. Индуктивность 300 мкГн.
Подключаем к аккумулятору:
Всего в первой серии опытов участвовало 9 аккумуляторов емкостью 55-75 а/час, возраст 1-5 лет.
Условия проведения опытов:
1. все аккумуляторы предварительно полностью разряжены и вновь заряжены током 0,1 от емкости аккумулятора, температура 20 гр.С, записано количество ампер-часов при зарядке (в зарядном устройстве есть счетчик ампер-часов).
2. импульсное устройство
(см. схему выше) подключается между аккумулятором и зарядным устройством
схему выше) подключается между аккумулятором и зарядным устройством
3. напряжение на выходе зарядного устройства стабилизировано =14,5В, энергия зарядного устройства расходуется на питание генератора импульсов и поддержание 14,5В на клеммах аккумулятора, т.е. аккумулятор в процессе десульфатации практически не заряжается и не разряжается
4. время десульфатации для каждого аккумулятора – 24 часа
5. сразу по окончании десульфатации измерена емкость аккумулятора: разряд током 0,1 от емкости аккумулятора, температура 20 гр.С, записано количество ампер-часов
Ток в импульсе для разных аккумуляторов был различным: изменялся от 3А до 810А, частота импульсов от 80 Гц (на больших токах) до 1500 Гц (на малых токах).
Осциллограммы напряжения на клеммах аккумулятора:
Максимальный ток в импульсе – 50А. Средний ток потребления по амперметру зарядного устройства =0,4А.
Максимальный ток в
импульсе – 810А. Средний ток потребления по амперметру зарядного устройства
=6,5А.
Ток импульсов измерялся осциллографом, как падение напряжения на 10 резисторах по 1 Ом, включенных параллельно. Суммарное сопротивление 0,1 Ом – для тока до 100А.
И на сопротивлении 0,01 Ом для токов более 100А.
Осциллограмма тока на резисторе 0,1 Ом:
Результаты:
Емкость всех аккумуляторов уменьшилась на 7-20%. Более новые аккумуляторы (1-2 года) пострадали меньше старых. Зависимости уменьшения емкости от паспортного значения (А/час) не выявлено, только от возраста. Падение емкости практически не зависит от амплитуды импульсов. Практически прямо пропорционально суммарной мощности импульсов: Pi х F, где
Pi – мощность одиночного импульса (пакета), F частота импульсов. Т.е. потеря емкости прямо пропорциональна суммарной энергии всех импульсов, поступивших на аккумулятор.
Меньше других потерял
емкость отечественный аккумулятор «SPUTNIK» 75а/час (возраст 1 год)
– 4,2%. От импортных собратьев отличается значительной массой – 20,6 кг.
От импортных собратьев отличается значительной массой – 20,6 кг.
Удручающие результаты, не правда ли?
Вторая серия опытов.
Нашел еще 3 импортных аккумулятора с аналогичными параметрами. Попробуем десульфатировать их «по старинке» с помощью этой схемы:
Условия десульфатации аналогичные:
1. все аккумуляторы предварительно полностью разряжены и вновь заряжены током 0,1 от емкости аккумулятора, температура 20 гр.С, записано количество ампер-часов при зарядке (в зарядном устройстве есть счетчик ампер-часов).
2. напряжение на клеммах аккумулятора стабилизировано =14,5В, лапочки автомобильные 12В 21Вт 2 шт., амперметр показывает средний ток =0, т.е. аккумулятор в процессе десульфатации практически не заряжается и не разряжается
3. время десульфатации для каждого аккумулятора – 24 часа
4. сразу по окончании десульфатации измерена емкость аккумулятора: разряд током 0,1 от емкости аккумулятора, температура 20 гр.С, записано количество ампер-часов
В этой схеме нет импульсов
повышенного напряжения. Первый полупериод (50 Гц) – заряд, второй – разряд. Осциллограммы
не делал.
Первый полупериод (50 Гц) – заряд, второй – разряд. Осциллограммы
не делал.
Результаты:
Емкость всех аккумуляторов уменьшилась на 2-2,5%.
Не так плохо, как в первой серии, но результат снова отрицательный. Мы ожидали увеличения емкости аккумулятора за счет снижения сульфатации пластин. Получилось наоборот. Очевидно, такой эффект может быть объяснен только одной причиной: импульсный ток интенсивно разрушает свинцовые пластины аккумуляторов.
Третья серия опытов.
К этому моменту я успел объехать все ближайшие автосервисы и пункты приема старых аккумуляторов J
Нашлось еще 2 аккумулятора.
Полностью разряжаем, затем заряжаем. Соблюдаем правила: разряд-заряд током 0,1 от емкости аккумулятора, температура 20 гр.С
Повторяем этот занимательный процесс 3 раза. И получаем положительный результат: емкость увеличилась на 24,5% и 31%.
Для примера: аккумулятор «Mutlu» красный, паспортная емкость 60А/час, возраст 3,5 года
Было 28,2А/час, стало 35,12А/час, прирост емкости 24,5%
Четвертая серия опытов.
Это делалось для самоуспокоения, типа «а вдруг не все потеряно?».
Взяли пару аккумуляторов, искалеченных в первой серии опытов и потренировали их, как третьей серии, но 4 цикла заряда-разряда. Удивительно: емкость увеличилась на 2% и 2,2%.
Выводы: современные аккумуляторы с тонкими пупырчато-губчатыми пластинами оказались совсем не прочными. Быстро разрушаются от импульсных токов, тем более от импульсов повышенного напряжения. Очевидно, так же быстро разрушаются от экстра-токов при включении стартера (запуск двигателя).
Старые тяжелые
аккумуляторы имели намного более толстые и гладкие пластины свинца. Очевидно,
сульфатация таких пластин то же наблюдалась, но в меньшей степени. Толстые и
гладкие пластины не боялись импульсных токов, намного легче десульфатировались.
При правильной эксплуатации имели большой срок службы. Для тех аккумуляторов
импульсные методы (устройства) десальфатации действительно могли быть панацеей. Для современных одноразовых аккумуляторов устройства десульфатации вредны.
Для современных одноразовых аккумуляторов устройства десульфатации вредны.
Рассказ знакомого: в деревне до сих пор эксплуатируется трактор 1937 года выпуска. С родным аккумулятором того же 1937 года. Хозяин регулярно снимает аккумулятор на подзарядку, проверяет уровень электролита, доливает дистиллированную воду. Зимой этот аккумулятор хранится в доме (в тепле).
Возможно, так оно и есть. Тем более, что свинец аккумуляторных пластин в те годы был не только значительно толще, но и чище (меньше примесей).
Асимметричная температурная модуляция для сверхбыстрой зарядки литий-ионных аккумуляторов
Основные характеристики
- •
Асимметричные температуры заряда и разряда обеспечивают длительную сверхбыструю зарядку
- •
Высокотемпературная зарядка устраняет гальванику за счет улучшенной транспортировки и кинетика
- •
Ограниченное время воздействия высокой температуры позволяет избежать резкого роста SEI
- •
Повышенная температура зарядки снижает потребность в охлаждении аккумулятора на> 12 ×
Контекст и масштаб
Электромобили будут только быть по-настоящему конкурентоспособными, если их можно заряжать так же быстро, как заправлять бензобак. Министерство энергетики США поставило цель разработать технологию экстремально быстрой зарядки (XFC), которая может увеличить запас хода на 200 миль за 10 минут. Критическим барьером для XFC является покрытие Li, которое обычно происходит при высоких скоростях заряда и резко снижает срок службы и безопасность аккумулятора. Здесь мы представляем метод асимметричной температурной модуляции (ATM), который заряжает литий-ионный элемент при повышенной температуре 60 ° C для устранения литиевого покрытия и ограничивает время воздействия 60 ° C только до 10 минут на цикл, чтобы предотвратить серьезные проблемы. деградация материалов.Используя промышленно доступные материалы для аккумуляторов, мы показываем, что литий-ионный аккумулятор с высокой энергией (209 Втч / кг), полученный методом банкомата, сохраняет емкость 91,7% после 2500 циклов XFC (равных 500000 миль пробега), что намного превышает показатели Департамента США. цели энергии (DOE) (500 циклов при 20% потере).
Министерство энергетики США поставило цель разработать технологию экстремально быстрой зарядки (XFC), которая может увеличить запас хода на 200 миль за 10 минут. Критическим барьером для XFC является покрытие Li, которое обычно происходит при высоких скоростях заряда и резко снижает срок службы и безопасность аккумулятора. Здесь мы представляем метод асимметричной температурной модуляции (ATM), который заряжает литий-ионный элемент при повышенной температуре 60 ° C для устранения литиевого покрытия и ограничивает время воздействия 60 ° C только до 10 минут на цикл, чтобы предотвратить серьезные проблемы. деградация материалов.Используя промышленно доступные материалы для аккумуляторов, мы показываем, что литий-ионный аккумулятор с высокой энергией (209 Втч / кг), полученный методом банкомата, сохраняет емкость 91,7% после 2500 циклов XFC (равных 500000 миль пробега), что намного превышает показатели Департамента США. цели энергии (DOE) (500 циклов при 20% потере).
Резюме
Добавление 200 миль за 10 минут, так называемая экстремально быстрая зарядка (XFC), является ключом к массовому внедрению аккумуляторных электромобилей (BEV). Здесь мы представляем метод асимметричной температурной модуляции (ATM), который, с одной стороны, заряжает литий-ионный элемент при повышенной температуре 60 ° C для устранения литиевого покрытия, а с другой стороны, ограничивает время воздействия при 60 ° C. только до ∼10 мин на цикл, или 0.1% от срока службы BEV, чтобы предотвратить серьезный рост межфазной границы твердого электролита. Асимметричная температура между зарядкой и разрядкой открывает новый путь для улучшения кинетики и транспортировки во время зарядки при сохранении длительного срока службы. Мы показываем, что элемент 9,5 Ач 170 Втч / кг выдержал 1700 циклов XFC (заряд 6 C до состояния заряда 80%) при 20% потере емкости с банкоматом, по сравнению с 60 циклами для контрольного элемента, и что 209 циклов -Ватт-час / кг BEV-элемент сохранил емкость 91,7% после 2500 циклов XFC.
Здесь мы представляем метод асимметричной температурной модуляции (ATM), который, с одной стороны, заряжает литий-ионный элемент при повышенной температуре 60 ° C для устранения литиевого покрытия, а с другой стороны, ограничивает время воздействия при 60 ° C. только до ∼10 мин на цикл, или 0.1% от срока службы BEV, чтобы предотвратить серьезный рост межфазной границы твердого электролита. Асимметричная температура между зарядкой и разрядкой открывает новый путь для улучшения кинетики и транспортировки во время зарядки при сохранении длительного срока службы. Мы показываем, что элемент 9,5 Ач 170 Втч / кг выдержал 1700 циклов XFC (заряд 6 C до состояния заряда 80%) при 20% потере емкости с банкоматом, по сравнению с 60 циклами для контрольного элемента, и что 209 циклов -Ватт-час / кг BEV-элемент сохранил емкость 91,7% после 2500 циклов XFC.
Ключевые слова
литий-ионный аккумулятор
экстремально быстрая зарядка
литиевое покрытие
твердый электролитный межфазный
высокотемпературный
терморегулятор
деградация аккумулятора
высокоэнергетический аккумулятор
Рекомендуемые статьи 0 © 2019 Elsevier Inc.
Рекомендуемые статьи
Ссылки на статьи
Battery Power Online | Асимметричная модуляция температуры улучшает чрезвычайно быструю зарядку литий-ионных батарей
От аккумулятора Интернет-персонал
30 октября 2019 г. | Для электромобилей станции быстрой зарядки считаются важным рыночным фактором. Возможность быстро подзарядить аккумуляторный электромобиль уменьшит «беспокойство» водителей о запасе хода. Доступны станции быстрой зарядки, но зарядка на 400 киловатт энергии или экстремально быстрая зарядка (XFC) могут увеличить дальность действия 200 миль за 10 минут.
Пока что для батареи, быстро потребляющей 400 киловатт энергии, существует риск литиевого покрытия (образование металлического лития вокруг анода), что серьезно сокращает срок службы батареи. Но исследователи из Университета штата Пенсильвания разработали литий-ионную батарею, которая заряжается при повышенной температуре, чтобы увеличить скорость реакции, но сохраняет батарею в прохладном состоянии во время разряда. Работа была опубликована сегодня в Джоулях (https://doi.org/10.1016/j.joule.2019.09.021).
Работа была опубликована сегодня в Джоулях (https://doi.org/10.1016/j.joule.2019.09.021).
Хотя обычные литиевые батареи заряжаются и разряжаются при одинаковой температуре, исследователи обнаружили, что проблему с литиевым покрытием можно обойти, зарядив батарею до повышенной температуры 60 градусов Цельсия в течение нескольких минут, а затем разрядив ее при более низких температурах.
«Ключевая идея, – пишут авторы, – состоит в том, чтобы решить дилемму между улучшенными характеристиками и ускоренной деградацией материалов при повышенных температурах с помощью метода асимметричной температурной модуляции (ATM). То есть элемент заряжается при высокой температуре, чтобы исключить покрытие из лития, и, с другой стороны, подвергается воздействию только в период быстрой зарядки, который составляет 10 минут за цикл. Поскольку рост SEI [твердый электролит-межфазный] зависит от времени, короткое время воздействия предотвращает фатальный рост SEI и, следовательно, эффективно контролирует деградацию клеток. ”
«В дополнение к быстрой зарядке эта конструкция позволяет нам ограничить время воздействия на аккумулятор повышенной температуры заряда, тем самым обеспечивая очень долгий срок службы», – говорит старший автор Чао-Ян Ван, инженер-механик из Университета штата Пенсильвания. «Главное – добиться быстрого нагрева; в противном случае аккумулятор будет оставаться при повышенных температурах слишком долго, что приведет к серьезной деградации ».
Чтобы сократить время нагрева и нагреть всю батарею до однородной температуры, Ван и его коллеги оснастили конструкцию литий-ионной батареи самонагревающейся никелевой структурой, которая нагревается менее чем за тридцать секунд.Чтобы протестировать свою модель, они зарядили три графитовых ячейки, предназначенные для гибридных электромобилей, при температуре 40, 49 и 60 градусов Цельсия, а также контрольную батарею при 20 градусах Цельсия, используя различные стратегии охлаждения для поддержания постоянной температуры заряда. Чтобы подтвердить, что литиевое покрытие не произошло, позже они полностью разрядили элементы и открыли их для анализа.
Ван и его команда обнаружили, что батареи, предварительно нагретые до 60 градусов Цельсия, могут выдерживать чрезвычайно быстрый процесс зарядки в течение 1700 циклов, в то время как контрольная ячейка может выдерживать только 60 циклов.При средней температуре заряда от 49 до 60 градусов Цельсия исследование не обнаружило никакого литиевого покрытия. Исследователи также заметили, что повышенная температура заряда значительно снижает охлаждение, необходимое для поддержания начальной температуры элемента – контрольный элемент генерирует 3,05 ватт-часа, в то время как элемент с температурой 60 градусов Цельсия генерирует только 1,7 ватт-часа.
«Раньше считалось, что ионно-литиевые батареи не должны работать при высоких температурах из-за опасности ускоренных побочных реакций», – говорит Ван.«Это исследование предполагает, что преимущества смягченного литиевого покрытия при повышенной температуре с ограниченным временем воздействия намного перевешивают негативное воздействие, связанное с обострением побочных реакций».
Исследователи отмечают, что технология полностью масштабируема, поскольку все ячейки основаны на промышленно доступных электродах; и они уже продемонстрировали его использование в крупномасштабных элементах, модулях и аккумуляторных батареях. Никелевая фольга увеличивает стоимость каждой ячейки на 0,47%, но поскольку конструкция устраняет необходимость во внешних нагревателях, используемых в текущих моделях, она фактически снижает стоимость производства каждой упаковки.
Далее команда Вана планирует сделать еще один шаг вперед в своем дизайне.
«Мы работаем над тем, чтобы зарядить энергоемкий аккумулятор электромобиля за пять минут, не повредив его», – говорит он. «Для этого потребуются высокостабильные электролиты и активные материалы в дополнение к самонагревающейся структуре, которую мы изобрели».
Печатные квазитвердотельные асимметричные суперконденсаторы с ионами магния для гибких интегрированных блоков с солнечной зарядкой
Конфигурация блоков зарядки от солнечных батарей
На Рисунке 1a (i) показана конфигурация гибкого блока с автономным питанием от солнечной энергии, включая модуль сбора энергии (т. е. гибкий солнечный элемент), модуль накопления энергии (то есть напечатанный квазитвердотельный массив ASC на полиимидной подложке) и закрывающий слой пластиковой пленки. Обратите внимание на рис. 1a (ii), что квазитвердотельный ASC собирается с использованием нитрида ванадия (VN) в качестве отрицательного электрода, оксида марганца (MnO 2 ) в качестве положительного электрода и MgSO 4 -полиакриламида. (ПАМ) гель в качестве электролита. Такое водное суперконденсаторное устройство работает на основе псевдоемкостной интеркаляции / деинтеркаляции ионов Mg 2+ между электродами VN и MnO 2 .Для интегрированного блока при воздействии солнечного света компонент солнечного элемента позволяет преобразовывать фотоизлучение в электричество и одновременно заряжать суперконденсатор (фотозарядка). Фотозаряжаемый суперконденсатор может обеспечивать накопленную мощность для электронных устройств в любое время по запросу (разрядка). Кроме того, вся интегрированная система имеет тонкопленочную планарную конструкцию, что придает ей хорошую гибкость.
е. гибкий солнечный элемент), модуль накопления энергии (то есть напечатанный квазитвердотельный массив ASC на полиимидной подложке) и закрывающий слой пластиковой пленки. Обратите внимание на рис. 1a (ii), что квазитвердотельный ASC собирается с использованием нитрида ванадия (VN) в качестве отрицательного электрода, оксида марганца (MnO 2 ) в качестве положительного электрода и MgSO 4 -полиакриламида. (ПАМ) гель в качестве электролита. Такое водное суперконденсаторное устройство работает на основе псевдоемкостной интеркаляции / деинтеркаляции ионов Mg 2+ между электродами VN и MnO 2 .Для интегрированного блока при воздействии солнечного света компонент солнечного элемента позволяет преобразовывать фотоизлучение в электричество и одновременно заряжать суперконденсатор (фотозарядка). Фотозаряжаемый суперконденсатор может обеспечивать накопленную мощность для электронных устройств в любое время по запросу (разрядка). Кроме того, вся интегрированная система имеет тонкопленочную планарную конструкцию, что придает ей хорошую гибкость. В качестве демонстрации концепции, автономный блок с солнечной зарядкой можно носить напрямую и работать как надежный источник питания для питания портативных электронных часов, как показано на рис.1а (iii).
Схематическое изображение интегрированного блока солнечной зарядки и характеристики нанопроволок VN. a Конфигурация устройства с автономным питанием от солнечной батареи и демонстрация концепции носимых устройств. b СЭМ-изображение готовых пористых нанопроволок VN. c ПЭМ-изображение нанопроволок ВН. Врезка: изображение СТЭМ высокого разрешения. d Рентгенограмма VN
Характеристика готовых нанопроволок VN
Синтетический путь получения нанопроволок VN был взят из описанного рецепта (дополнительный рис.1а) 8 . Изображения, полученные с помощью сканирующей электронной микроскопии (SEM; рис. 1b), показывают типичную пористую структуру нанопроволоки VN с однородной шириной 120–150 нм и длиной ~ 2 мкм. Просвечивающая электронная микроскопия (ПЭМ; рис. 1с) представляет подробную морфологию пористых нанопроволок VN, показывая множество взаимосвязанных открытых пор. Элементное картирование свежеприготовленного VN показывает однородное распределение азота и ванадия (дополнительный рис. 1b). Появление кислородного сигнала свидетельствует о существовании поверхностно связанного кислорода при контакте с воздухом.Дальнейший анализ XPS указывает на преобладание состояния связывания V 3+ , а также сигнала V-N (дополнительный рис. 1c, d), что подразумевает успешное изготовление VN. Обратите внимание, что отсутствуют пики XPS, указывающие на состояния V 4+ и V 5+ , что может быть связано с чрезмерным восстановлением в результате длительного и высокотемпературного процесса отжига NH 3 (600 ° C для 3 ч). Наблюдения с помощью сканирующей просвечивающей электронной микроскопии (STEM) с коррекцией аберрации отображают полосы решетки VN (рис.1с вставка; Дополнительный рис.
 1e – g) с шагом решетки 0,21 нм, хорошо совпадающим с плоскостью VN (200). На рисунке 1d представлена рентгенограмма синтезированных образцов VN. Характерные пики находятся при 37,9 ° (111), 44,0 ° (200) и 64,1 ° (220), подтверждая четко определенную кристаллическую структуру VN (JCPDS № 25-1252). Ожидается, что такой VN в форме нанопроволоки является ключом к достижению благоприятных электрохимических характеристик при использовании в качестве электродов из-за: (1) большого аспектного отношения, собственной пористой структуры и размеров нанометрового размера (<20 нм) нанопроволок VN, которые обеспечивают они обладают высокоскоростными возможностями и приповерхностными псевдоемкостными интеркаляционными свойствами Mg 2+ , что является решающим фактором для их превосходных скоростных характеристик и стабильности при циклическом воздействии.(2) микроструктуры в форме нанопроволоки могут переплетаться друг с другом, тем самым избегая агрегации при циклировании, что часто наблюдается при использовании морфологий наночастиц или нанолистов.
1e – g) с шагом решетки 0,21 нм, хорошо совпадающим с плоскостью VN (200). На рисунке 1d представлена рентгенограмма синтезированных образцов VN. Характерные пики находятся при 37,9 ° (111), 44,0 ° (200) и 64,1 ° (220), подтверждая четко определенную кристаллическую структуру VN (JCPDS № 25-1252). Ожидается, что такой VN в форме нанопроволоки является ключом к достижению благоприятных электрохимических характеристик при использовании в качестве электродов из-за: (1) большого аспектного отношения, собственной пористой структуры и размеров нанометрового размера (<20 нм) нанопроволок VN, которые обеспечивают они обладают высокоскоростными возможностями и приповерхностными псевдоемкостными интеркаляционными свойствами Mg 2+ , что является решающим фактором для их превосходных скоростных характеристик и стабильности при циклическом воздействии.(2) микроструктуры в форме нанопроволоки могут переплетаться друг с другом, тем самым избегая агрегации при циклировании, что часто наблюдается при использовании морфологий наночастиц или нанолистов.
Исследование электрохимических свойств нанопроволок VN
Электрохимические характеристики VN систематически исследовались с помощью системы трехэлектродных ячеек. Измерения циклической вольтамперометрии (ЦВА) при скорости сканирования 10 мВ с -1 проводили в различных нейтральных водных электролитах с одинаковыми концентрациями катионов (рис.2а), в том числе 0,5 M Li 2 SO 4 , 0,5 M Na 2 SO 4 , 0,5 M K 2 SO 4 и 1,0 M MgSO 4 . Интересно отметить, что электрод VN имеет самую большую площадь CV в ионном электролите Mg. На рисунке 2b приведены значения удельных емкостей, рассчитанных на основе профилей CV. Электрод VN демонстрирует удельную емкость 230 Ф · г -1 в электролите MgSO 4 , что намного превосходит значения, достигнутые в Li 2 SO 4 (120 Ф · г -1 ), Na 2 SO 4 (100 F g -1 ) и K 2 SO 4 (70 F g -1 ). – \ leftrightarrow {\ mathrm {VN}} _ {{x}} {\ mathrm {O}} _ {{y}} {\ mathrm {Mg}} _ {{z}}. $$
(1)
Рис. 2Электрохимические характеристики ВН в трехэлектродной конфигурации. a Кривые CV, измеренные в различных электролитах на основе катионов при одинаковой скорости сканирования 10 мВ с -1 . b Соответствующие удельные гравиметрические емкости, рассчитанные из a в различных электролитах. c Поляризационные кривые электрода VN в различных нейтральных электролитах на основе катионов. d Удельные емкости VN в 1,0 M электролите MgSO 4 при различных скоростях сканирования. Вставка: соответствующие кривые CV при разных скоростях сканирования. e Емкостные и контролируемые диффузией вклады в общее накопление заряда VN в 1,0 M MgSO 4 при 10 мВ с −1 . f Нормализованное соотношение вкладов емкостной (голубой) и контролируемой диффузией (оранжевый) емкости при разных скоростях сканирования
В этом отношении емкость, хранящаяся в системе Mg 2+ , должна быть вдвое больше, чем у других систем одновалентных катионов, что хорошо согласуется с емкостью, полученной из измерений CV. (ii) Относительно меньший ионный радиус ионов Mg 2+ (Mg 2+ : 0,72 Å, Li + : 0,76 Å, Na + : 1,02 Å, K + : 1,51 Å) 10 приводит к псевдо-емкостному поведению накопления заряда электрода VN.
Исследования с помощью спектроскопии электрохимического импеданса (EIS) предлагают более глубокое понимание ионных и электронных транспортных свойств VN-электрода в различных катионных электролитах (дополнительный рис. 2a, b). В низкочастотной области крутая форма прямых линий показывает типичное емкостное поведение в различных электролитах.В высокочастотной области внутреннее сопротивление ( R с ) показывает очевидные различия (K + : 4 Ом, Na + : 5,5 Ом, Li + : 7 Ом, Mg 2+ : 8,5 Ом). R s обычно состоит из проводящего сопротивления электролита, а также собственного контактного сопротивления между материалом электрода и токосъемником. Поскольку все графики Найквиста были получены на основе одного и того же материала электрода и конфигурации электрохимической ячейки, различие R s следует в основном отнести к разным проводящим сопротивлениям различных катионных электролитов.Из-за своей двухвалентной природы и небольшого ионного радиуса ионы Mg 2+ обладают относительно прочными ионными связями. Следовательно, диффузия ионов Mg 2+ более медленная, чем диффузия одновалентных катионов. Поляризация более высокого сопротивления Mg 2+ должна приводить к большему гистерезису напряжения, что помогает увеличить стабильное рабочее напряжение VN в системе электролита Mg 2+ . Приведена эквивалентная схема Рэндлса с относительными параметрами (дополнительный рис.2c; Дополнительная таблица 1), сопровождаемая подробным обсуждением, показанным в дополнительном примечании 1. На рисунке 2c показаны поляризационные кривые линейной вольтамперометрии (LSV) для электрода VN в различных катионных электролитах.
Поскольку все графики Найквиста были получены на основе одного и того же материала электрода и конфигурации электрохимической ячейки, различие R s следует в основном отнести к разным проводящим сопротивлениям различных катионных электролитов.Из-за своей двухвалентной природы и небольшого ионного радиуса ионы Mg 2+ обладают относительно прочными ионными связями. Следовательно, диффузия ионов Mg 2+ более медленная, чем диффузия одновалентных катионов. Поляризация более высокого сопротивления Mg 2+ должна приводить к большему гистерезису напряжения, что помогает увеличить стабильное рабочее напряжение VN в системе электролита Mg 2+ . Приведена эквивалентная схема Рэндлса с относительными параметрами (дополнительный рис.2c; Дополнительная таблица 1), сопровождаемая подробным обсуждением, показанным в дополнительном примечании 1. На рисунке 2c показаны поляризационные кривые линейной вольтамперометрии (LSV) для электрода VN в различных катионных электролитах. Очевидно, что система Mg 2+ проявляет наибольший перенапряжение по выделению водорода. В соответствии с этим можно увеличить стабильное рабочее напряжение электрода VN в электролите Mg 2+ , что подтверждает анализ EIS. На рисунке 2d приведены удельные емкости VN в электролите Mg 2+ , которые соответствуют профилям CV при разных скоростях сканирования (рис.2г, врезка). Формы CV хорошо сохраняются при увеличении скорости сканирования с 1 до 200 мВ с -1 , что указывает на благоприятные скоростные характеристики электрода VN в системе Mg 2+ . Обратите внимание, что пик окислительно-восстановительного потенциала между -0,6 и -0,7 В показывает только небольшой сдвиг при увеличении скорости сканирования, что указывает на легкую кинетику реакции во время накопления заряда. Таким образом, заряд, накопленный вкладом с управляемой диффузией или нефарадеевской емкостью, может быть проанализирован с помощью значения b , которое может быть получено на основе уравнения i = aν b 11 .
Очевидно, что система Mg 2+ проявляет наибольший перенапряжение по выделению водорода. В соответствии с этим можно увеличить стабильное рабочее напряжение электрода VN в электролите Mg 2+ , что подтверждает анализ EIS. На рисунке 2d приведены удельные емкости VN в электролите Mg 2+ , которые соответствуют профилям CV при разных скоростях сканирования (рис.2г, врезка). Формы CV хорошо сохраняются при увеличении скорости сканирования с 1 до 200 мВ с -1 , что указывает на благоприятные скоростные характеристики электрода VN в системе Mg 2+ . Обратите внимание, что пик окислительно-восстановительного потенциала между -0,6 и -0,7 В показывает только небольшой сдвиг при увеличении скорости сканирования, что указывает на легкую кинетику реакции во время накопления заряда. Таким образом, заряд, накопленный вкладом с управляемой диффузией или нефарадеевской емкостью, может быть проанализирован с помощью значения b , которое может быть получено на основе уравнения i = aν b 11 . В этом смысле значение b рассчитывается соответственно при 0,91 и 0,90 для анодного и катодного пиков (дополнительный рис. 2d), что означает, что вклад емкостного поведения преобладает. Чтобы дополнительно идентифицировать соотношение вкладов, текущий отклик может быть выражен путем объединения двух отдельных механизмов (поведение поверхностной емкости и контролируемые диффузией процессы введения Mg 2+ ): i (V) = k 1 ν + k 2 ν 1/2 .Здесь k 1 ν и k 2 ν 1/2 представляют вклады тока, контролируемые емкостью и диффузией, соответственно. На рисунке 2e показан вклад емкостного тока при CV-сканировании 10 мВ с -1 , демонстрирующий большой процент емкости (72,6%). Кроме того, на рис. 2е представлена гистограмма рассчитанных емкостных вкладов при различных скоростях сканирования.
В этом смысле значение b рассчитывается соответственно при 0,91 и 0,90 для анодного и катодного пиков (дополнительный рис. 2d), что означает, что вклад емкостного поведения преобладает. Чтобы дополнительно идентифицировать соотношение вкладов, текущий отклик может быть выражен путем объединения двух отдельных механизмов (поведение поверхностной емкости и контролируемые диффузией процессы введения Mg 2+ ): i (V) = k 1 ν + k 2 ν 1/2 .Здесь k 1 ν и k 2 ν 1/2 представляют вклады тока, контролируемые емкостью и диффузией, соответственно. На рисунке 2e показан вклад емкостного тока при CV-сканировании 10 мВ с -1 , демонстрирующий большой процент емкости (72,6%). Кроме того, на рис. 2е представлена гистограмма рассчитанных емкостных вкладов при различных скоростях сканирования. Очевидно, что отношение емкостного вклада увеличивается (с 60% до 98%) по мере увеличения скорости сканирования с 1 до 100 мВ с -1 , дополнительно подтверждая преобладающий емкостной механизм реакции электрода VN.
Исследование механизма электрохимической реакции VN
Для изучения механизма реакции ионов Mg 2+ на электроде VN был проведен операндный XRD-анализ с использованием VN в качестве целевого электрода и MnO 2 в качестве противоэлектрода. . Все рентгенограммы были собраны во втором цикле после зарядки / разрядки, чтобы избежать необратимых реакций, которые произошли во время первого цикла. Обратите внимание, что в течение всего процесса зарядки / разрядки новых фаз не обнаруживается.На рис. 3а показан очевидный сдвиг пика для плоскости VN (200) (2 θ : 44–45 °). При зарядке (0–2,2 В) дифракционный пик постепенно смещается в сторону больших углов (2 θ : 44,5–44,7 °), указывая на сокращение межплоскостного расстояния в направлении (200). В ответ дифракционный пик возвращается в низкоугловое положение при разрядке, указывая на расширение интервала решетки. Межплоскостное расстояние рассчитывается по уравнению Брэгга ( λ = 0.15406 нм), чтобы обратимо увеличиваться на 0,2 Å (2,1 → 1,9 → 2,1 Å) в процессе заряда / разряда (как показано на рис. 3b). Такое периодическое изменение шага решетки подтверждает обратимую псевдоемкостную интеркаляцию / деинтеркаляцию иона Mg плоскости VN (200), не вызывая фазового перехода 11 . В то же время сокращение межплоскостного расстояния во время псевдоемкостной интеркаляции ионов Mg можно объяснить увеличением электростатического притяжения между гостевыми ионами Mg и основной решеткой VN 12,13,14 .
В ответ дифракционный пик возвращается в низкоугловое положение при разрядке, указывая на расширение интервала решетки. Межплоскостное расстояние рассчитывается по уравнению Брэгга ( λ = 0.15406 нм), чтобы обратимо увеличиваться на 0,2 Å (2,1 → 1,9 → 2,1 Å) в процессе заряда / разряда (как показано на рис. 3b). Такое периодическое изменение шага решетки подтверждает обратимую псевдоемкостную интеркаляцию / деинтеркаляцию иона Mg плоскости VN (200), не вызывая фазового перехода 11 . В то же время сокращение межплоскостного расстояния во время псевдоемкостной интеркаляции ионов Mg можно объяснить увеличением электростатического притяжения между гостевыми ионами Mg и основной решеткой VN 12,13,14 .
Механизм электрохимической реакции ВН в нейтральном электролите MgSO 4 . a Рентгенограммы Operando XRD VN в процессе заряда / разряда (плотность тока: 0,1 А г -1 ). b Схематическая диаграмма, показывающая сжатие и расширение кристаллической плоскости (200) во время процесса заряда / разряда. c Ex situ XPS Mg 1 s спектры при различных потенциалах во время CV-сканирования при скорости сканирования 5 мВ с -1 . d Изменение валентного состояния ванадия по данным XPS V 2p-спектров
c Ex situ XPS Mg 1 s спектры при различных потенциалах во время CV-сканирования при скорости сканирования 5 мВ с -1 . d Изменение валентного состояния ванадия по данным XPS V 2p-спектров
Псевдоемкостный механизм интеркаляции ионов Mg VN-электрода был подтвержден ex situ Mg 1 s XPS-измерениями. Как показано на фиг. 3c, семь различных точек выборки взяты из CV-сканирования со скоростью сканирования 5 мВ с -1 . Пик Mg 1 s центрирован при 1303,3 эВ, что может быть отнесено к Mg 2+ . Что касается процесса зарядки (переход от точки 1 к 5), можно четко наблюдать постепенное увеличение интенсивности пика, отражающее интеркаляцию ионов Mg 2+ .Во время процесса разряда (от -0,9 до 0 В) снижение интенсивности сигнала означает деинтеркаляцию Mg 2+ . Кроме того, ex situ V 2 p XPS-спектры показывают изменение валентного состояния ванадия при зарядке / разряде (дополнительный рис. 3). Пики при 515,4 и 513,2 эВ представляют вклад от V 3+ и V 2+ , соответственно, 15 , без присутствия каких-либо новых сигналов. В этом отношении можно сделать вывод, что V 3+ является стабильным состоянием валентности окисления в этом электроде VN, тогда как V 2+ является относительно стабильным состоянием валентности восстановления.Стоит отметить, что компенсация заряда будет происходить, когда количество положительного заряда в активных материалах увеличивается 16 ; Другими словами, при внедрении ионов Mg 2+ в решетку VN. На рисунке 3d показана доля валентных состояний ванадия из спектров ex situ V 2p. В исходном состоянии концентрация V 3+ и V 2+ составляет 64% и 36% соответственно (точка 1). В свою очередь, соотношение V 3+ уменьшается до 58% в полностью заряженном состоянии (точка 5).После полной разрядки доля V 3+ / V 2+ возвращается к 63% / 37% (точка 7), что соответствует исходному состоянию.
3). Пики при 515,4 и 513,2 эВ представляют вклад от V 3+ и V 2+ , соответственно, 15 , без присутствия каких-либо новых сигналов. В этом отношении можно сделать вывод, что V 3+ является стабильным состоянием валентности окисления в этом электроде VN, тогда как V 2+ является относительно стабильным состоянием валентности восстановления.Стоит отметить, что компенсация заряда будет происходить, когда количество положительного заряда в активных материалах увеличивается 16 ; Другими словами, при внедрении ионов Mg 2+ в решетку VN. На рисунке 3d показана доля валентных состояний ванадия из спектров ex situ V 2p. В исходном состоянии концентрация V 3+ и V 2+ составляет 64% и 36% соответственно (точка 1). В свою очередь, соотношение V 3+ уменьшается до 58% в полностью заряженном состоянии (точка 5).После полной разрядки доля V 3+ / V 2+ возвращается к 63% / 37% (точка 7), что соответствует исходному состоянию. Учитывая поверхностно-чувствительный характер метода XPS, эти результаты показывают, что сильно обратимые окислительно-восстановительные реакции на поверхности между V 3+ / V 2+ также вносят вклад в механизм накопления заряда во время электрохимического процесса.
Учитывая поверхностно-чувствительный характер метода XPS, эти результаты показывают, что сильно обратимые окислительно-восстановительные реакции на поверхности между V 3+ / V 2+ также вносят вклад в механизм накопления заряда во время электрохимического процесса.
Оценка характеристик квазитвердотельных ASC с ионами Mg
Квазитвердотельные ASC были собраны в типичных конфигурациях плоских элементов, в которых композит MnO 2 @carbon (MnO 2 @C) использовался в качестве положительный электрод для введения / извлечения ионов Mg 2+ .Синтезированные композиты были подвергнуты широкому набору инструментов определения характеристик (дополнительный рис. 4). Трехмерная наноархитектура MnO 2 усиливает диффузию ионов Mg, в то время как проводящая углеродная матрица улучшает электропроводность гибридного электрода. Электрохимические характеристики композитов MnO 2 @C систематически измерялись в трехэлектродной системе (дополнительный рис. 5). Электрод обеспечивает удельную емкость 240 Ф · г -1 при скорости сканирования 10 мВ · с -1 с широким потенциалом окна (от -0.От 3 до 1,2 В относительно Ag / AgCl), что аналогично другим системам катионных электролитов (Li + , Na + и K + ). Электрод MnO 2 , вероятно, будет проявлять механизм накопления заряда с преобладанием твердотельной диффузии, который отличается от поведения емкостного накопления заряда электрода VN (дополнительный рисунок 6) 17 .
5). Электрод обеспечивает удельную емкость 240 Ф · г -1 при скорости сканирования 10 мВ · с -1 с широким потенциалом окна (от -0.От 3 до 1,2 В относительно Ag / AgCl), что аналогично другим системам катионных электролитов (Li + , Na + и K + ). Электрод MnO 2 , вероятно, будет проявлять механизм накопления заряда с преобладанием твердотельной диффузии, который отличается от поведения емкостного накопления заряда электрода VN (дополнительный рисунок 6) 17 .
Чтобы собрать квазитвердотельный ASC, гель крахмал / ПАМ / MgSO 4 был введен в качестве электролита 18 .По сравнению с обычно используемым поливиниловым спиртом такая гелевая матрица на основе ПАМ демонстрирует превосходную совместимость с катионами Mg. Как и ожидалось, молекулы крахмала и молекулы акриламида физически переплетаются и взаимопроникают друг с другом, образуя ионопроводящую сеть с хорошей водопоглощающей способностью и механической прочностью, как показано на рис. 4а. Свежеприготовленный гель-электролит может вернуться в исходное состояние после различных деформаций, таких как сплющивание, изгиб и / или растяжение.На рисунке 4b представлены кривые CV как для отрицательного электрода VN, так и для положительного электрода из MnO 2 при скорости сканирования 10 мВ с -1 , что указывает на то, что сконструированное асимметричное устройство может достичь окна напряжения 2,1 В или даже больше. Затем были выполнены CV-сканирование в различных диапазонах напряжения от 1,4 до 2,6 В. Обратите внимание, что явной поляризации нет даже при увеличении приложенного напряжения до 2,2 В (рис. 4c, красная линия). Такой широкий диапазон напряжений превосходит большинство недавно представленных ASC 19,20 .На рисунке 4d показаны кривые гальваностатического заряда / разряда (GCD) наших квазитвердотельных ASC при различных плотностях тока от 2 до 12 мА · см 90 · 107 −2 90 · 108. При 2 мА · см 90 · 107 −2 90 · 108 время разряда достигает 637 с, что соответствует поверхностной емкости 576 мФ · см 90 · 107 −2 90 · 108, что составляет 79,2% от идентичного устройства, испытанного в жидком электролите (рис.
4а. Свежеприготовленный гель-электролит может вернуться в исходное состояние после различных деформаций, таких как сплющивание, изгиб и / или растяжение.На рисунке 4b представлены кривые CV как для отрицательного электрода VN, так и для положительного электрода из MnO 2 при скорости сканирования 10 мВ с -1 , что указывает на то, что сконструированное асимметричное устройство может достичь окна напряжения 2,1 В или даже больше. Затем были выполнены CV-сканирование в различных диапазонах напряжения от 1,4 до 2,6 В. Обратите внимание, что явной поляризации нет даже при увеличении приложенного напряжения до 2,2 В (рис. 4c, красная линия). Такой широкий диапазон напряжений превосходит большинство недавно представленных ASC 19,20 .На рисунке 4d показаны кривые гальваностатического заряда / разряда (GCD) наших квазитвердотельных ASC при различных плотностях тока от 2 до 12 мА · см 90 · 107 −2 90 · 108. При 2 мА · см 90 · 107 −2 90 · 108 время разряда достигает 637 с, что соответствует поверхностной емкости 576 мФ · см 90 · 107 −2 90 · 108, что составляет 79,2% от идентичного устройства, испытанного в жидком электролите (рис. 4e; дополнительный рис. . 7). Интересно, что когда плотности тока превышают 4 мА · см 90 · 107 −2 90 · 108, поверхностная емкость гелевого электролита становится даже выше, чем у системы на основе жидкого электролита.Превосходная производительность гелевого электролита по сравнению с жидким электролитом заслуживает дальнейшего исследования. Таким образом, были собраны сравнительные профили EIS между гелевым электролитом и системами жидкого электролита до и после электрохимических испытаний (рис. 4f; дополнительный рис. 8). Все эти графики Найквиста соответствуют эквивалентной схеме Рэндлса (дополнительная таблица 2). Очевидно, что R s гелевой электролитной системы демонстрирует лишь незначительное увеличение (0.037 Ом) после электрохимических измерений, что меньше 0,243 Ом системы жидкого электролита. Более крутая линия в низкочастотном диапазоне на графиках Найквиста для гелевой системы (особенно после электрохимических испытаний) также указывает на усиление диффузии ионов.
4e; дополнительный рис. . 7). Интересно, что когда плотности тока превышают 4 мА · см 90 · 107 −2 90 · 108, поверхностная емкость гелевого электролита становится даже выше, чем у системы на основе жидкого электролита.Превосходная производительность гелевого электролита по сравнению с жидким электролитом заслуживает дальнейшего исследования. Таким образом, были собраны сравнительные профили EIS между гелевым электролитом и системами жидкого электролита до и после электрохимических испытаний (рис. 4f; дополнительный рис. 8). Все эти графики Найквиста соответствуют эквивалентной схеме Рэндлса (дополнительная таблица 2). Очевидно, что R s гелевой электролитной системы демонстрирует лишь незначительное увеличение (0.037 Ом) после электрохимических измерений, что меньше 0,243 Ом системы жидкого электролита. Более крутая линия в низкочастотном диапазоне на графиках Найквиста для гелевой системы (особенно после электрохимических испытаний) также указывает на усиление диффузии ионов. Такая лучшая способность к диффузии ионов электролита является ключом к достижению более высоких скоростных характеристик. Между тем, исчерпывающая морфологическая и элементная характеристика свидетельствует о том, что уникальная пористая структура, высокое соотношение содержания воды и плотная граница раздела электролит / электрод способствуют улучшенным характеристикам диффузии ионов системы на основе гелевого электролита (дополнительные рис.9–12). Примечательно, что сохранение емкости 95% может быть достигнуто при высокой плотности тока 16 мА · см 90 · 107 −2 90 · 108 после 5000 циклов, что демонстрирует выдающуюся циклическую стабильность собранного квазитвердотельного ASC (рис. 4g). Кроме того, нет искажения кривых заряда / разряда даже для последних нескольких циклов, как показано на вставке к рис. 4g.
Такая лучшая способность к диффузии ионов электролита является ключом к достижению более высоких скоростных характеристик. Между тем, исчерпывающая морфологическая и элементная характеристика свидетельствует о том, что уникальная пористая структура, высокое соотношение содержания воды и плотная граница раздела электролит / электрод способствуют улучшенным характеристикам диффузии ионов системы на основе гелевого электролита (дополнительные рис.9–12). Примечательно, что сохранение емкости 95% может быть достигнуто при высокой плотности тока 16 мА · см 90 · 107 −2 90 · 108 после 5000 циклов, что демонстрирует выдающуюся циклическую стабильность собранного квазитвердотельного ASC (рис. 4g). Кроме того, нет искажения кривых заряда / разряда даже для последних нескольких циклов, как показано на вставке к рис. 4g.
Электрохимические характеристики квазитвердотельных ИСК с ионами Mg. a Структурная схема гелевого электролита ПАМ и цифровые фотографии, показывающие его механическую прочность при деформации. b Кривая CV, полученная для электродов VN и MnO 2 , соответственно, в различных диапазонах потенциалов при скорости сканирования 10 мВ с -1 . c CV-кривые квазитвердотельного ASC с увеличивающимся окном напряжения от 1,4 до 2,6 В при 10 мВ с -1 . d Кривые гальваностатического заряда / разряда ASC при различной плотности тока. e Сравнение удельной емкости жидкого электролита и гелевого электролита при различных плотностях тока. f График Найквиста жидкого электролита / сепаратора и гелевого электролита. г Долговременная циклическая стабильность квазитвердотельного ASC при плотности тока заряда / разряда 16 мА см −2 . h График Рэгона наших квазитвердотельных устройств ASC в сравнении с другими недавно опубликованными квазитвердотельными устройствами ASC в нейтральных электролитах
b Кривая CV, полученная для электродов VN и MnO 2 , соответственно, в различных диапазонах потенциалов при скорости сканирования 10 мВ с -1 . c CV-кривые квазитвердотельного ASC с увеличивающимся окном напряжения от 1,4 до 2,6 В при 10 мВ с -1 . d Кривые гальваностатического заряда / разряда ASC при различной плотности тока. e Сравнение удельной емкости жидкого электролита и гелевого электролита при различных плотностях тока. f График Найквиста жидкого электролита / сепаратора и гелевого электролита. г Долговременная циклическая стабильность квазитвердотельного ASC при плотности тока заряда / разряда 16 мА см −2 . h График Рэгона наших квазитвердотельных устройств ASC в сравнении с другими недавно опубликованными квазитвердотельными устройствами ASC в нейтральных электролитах
На рисунке 4h представлен график Рагона объемной плотности энергии и плотности мощности наших квазитвердотельных устройств. твердотельные ASC (VN // MnO 2 ) по сравнению с другими недавно опубликованными квазитвердотельными суперконденсаторами, испытанными в нейтральных гелевых электролитах.Наше устройство демонстрирует объемную плотность энергии 13,1 мВтч см −3 при плотности мощности 72 мВт см −3 . Он также сохраняет 79% своей плотности энергии при увеличении плотности мощности до 440 мВт / см −3 . Эти значения заметно превосходят показатели современных систем 9,19,20,21,22,23,24,25,26,27,28 , включая VO x // VN (LiCl / PVA) 9 , MnO 2 @ NG // AC (LiCl / PVA) 24 , VN @ MOF // AC (Na 2 SO 4 / PVA) 19 и V 2 O 3 @ C // Fe 3 O 4 @ TiO 2 (LiCl / PVA) 26 .Высокая плотность энергии и отличная производительность нашего устройства в основном объясняются значительно расширенным стабильным диапазоном напряжений ионной системы Mg и благоприятным псевдоемкостным накоплением заряда ионов Mg отрицательного электрода VN.
твердотельные ASC (VN // MnO 2 ) по сравнению с другими недавно опубликованными квазитвердотельными суперконденсаторами, испытанными в нейтральных гелевых электролитах.Наше устройство демонстрирует объемную плотность энергии 13,1 мВтч см −3 при плотности мощности 72 мВт см −3 . Он также сохраняет 79% своей плотности энергии при увеличении плотности мощности до 440 мВт / см −3 . Эти значения заметно превосходят показатели современных систем 9,19,20,21,22,23,24,25,26,27,28 , включая VO x // VN (LiCl / PVA) 9 , MnO 2 @ NG // AC (LiCl / PVA) 24 , VN @ MOF // AC (Na 2 SO 4 / PVA) 19 и V 2 O 3 @ C // Fe 3 O 4 @ TiO 2 (LiCl / PVA) 26 .Высокая плотность энергии и отличная производительность нашего устройства в основном объясняются значительно расширенным стабильным диапазоном напряжений ионной системы Mg и благоприятным псевдоемкостным накоплением заряда ионов Mg отрицательного электрода VN.
Создание гибких микро-асимметричных суперконденсаторов
В поисках портативных, миниатюрных и носимых устройств накопления энергии мы затем изготовили гибкие микро-асимметричные суперконденсаторы (MASC), состоящие из VN и MnO 2 встречно-штыревых электродов посредством трафаретной печати 29 , 31,32 (дополнительный рис.13). На рисунке 5a изображена общая процедура прямой печати MASC, включая предварительное формирование рисунка из пленок Au в качестве токосъемников 33 , с последующим нанесением встречно-штыревых электродов VN и MnO 2 и нанесением PAM-MgSO. 4 Гель-электролит (подробные размеры рисунка трафаретной печати показаны на дополнительном рис. 14). Электрохимические характеристики сконструированных MASC VN // MnO 2 были охарактеризованы измерениями CV и GCD.На рисунке 5b показаны CV-профили MASC при разных скоростях сканирования. Обратите внимание, что напряжение холостого хода достигает впечатляющих 2,2 В, превышая большинство недавно зарегистрированных микроконденсаторов и ASC. Пара пиков окислительно-восстановительного потенциала все еще может быть обнаружена, что хорошо согласуется с приведенными выше результатами квазитвердотельного асимметричного SC в конфигурации плоской ячейки. Форма CV хорошо сохраняется в диапазоне от 10 до 200 мВ с -1 , что свидетельствует о превосходных скоростных характеристиках устройства. На рисунке 5c показаны кривые GCD при различных плотностях тока от 0.От 5 до 12 мА · см −2 , демонстрируя очевидные треугольные формы. Таким образом, наш напечатанный MASC обеспечивает поверхностную емкость 28,5 мФ см −2 при 0,5 мА см −2 , в то время как 42% этой емкости остается после тестирования при 12 мА см −2 , как показано на рис. 5г. Полученные таким образом MASC демонстрируют медленный процесс саморазряда (дополнительный рисунок 15) и стабильную работу при длительном цикле (сохранение емкости 90% после 8000 циклов; см. Дополнительный рисунок 16). Кроме того, график Рагона, изображенный на дополнительном рис.
Пара пиков окислительно-восстановительного потенциала все еще может быть обнаружена, что хорошо согласуется с приведенными выше результатами квазитвердотельного асимметричного SC в конфигурации плоской ячейки. Форма CV хорошо сохраняется в диапазоне от 10 до 200 мВ с -1 , что свидетельствует о превосходных скоростных характеристиках устройства. На рисунке 5c показаны кривые GCD при различных плотностях тока от 0.От 5 до 12 мА · см −2 , демонстрируя очевидные треугольные формы. Таким образом, наш напечатанный MASC обеспечивает поверхностную емкость 28,5 мФ см −2 при 0,5 мА см −2 , в то время как 42% этой емкости остается после тестирования при 12 мА см −2 , как показано на рис. 5г. Полученные таким образом MASC демонстрируют медленный процесс саморазряда (дополнительный рисунок 15) и стабильную работу при длительном цикле (сохранение емкости 90% после 8000 циклов; см. Дополнительный рисунок 16). Кроме того, график Рагона, изображенный на дополнительном рис. 17 демонстрирует, что наши напечатанные MASC собирают плотность энергии 19,13 мкВтч см −2 при плотности мощности 0,55 мВт см −2 , что превосходит таковые у недавно зарегистрированных плоских микроконденсаторов (подробную информацию см. В дополнительной таблице 3). ).
17 демонстрирует, что наши напечатанные MASC собирают плотность энергии 19,13 мкВтч см −2 при плотности мощности 0,55 мВт см −2 , что превосходит таковые у недавно зарегистрированных плоских микроконденсаторов (подробную информацию см. В дополнительной таблице 3). ).
Электрохимические характеристики гибкого и пригодного для печати квазитвердотельного MASC. a Схематическое изображение процесса изготовления MgSO 4 / PAM VN // MnO 2 MASC. b CV-кривые напечатанного MASC, полученные при различных скоростях сканирования. c Кривые гальваностатического заряда / разряда, испытанные при различных плотностях тока. d Расчетная удельная емкость при различных плотностях тока, соответствующая c . e Кривые гальваностатического заряда / разряда, полученные при 0,5 мА см –2 одиночного, последовательного и параллельного соединения двух MASC. f Кривые гальваностатического заряда / разряда протестированы при 0.5 мА см −2 под разными углами изгиба
f Кривые гальваностатического заряда / разряда протестированы при 0.5 мА см −2 под разными углами изгиба
Для эффективного повышения рабочего напряжения и выходной емкости одного устройства MASC можно легко подключить последовательно или параллельно за счет оптимизации процесса печати. На рис. 5д представлены кривые НОД одиночных, последовательных и параллельных MASC. В этом отношении последовательное соединение позволяет увеличить выходное напряжение до 4,4 В (вдвое больше 2,2 В для одного устройства). Между тем, емкость можно легко удвоить, используя параллельное соединение, демонстрируя расширенную модуляцию для интеграции устройства 33 .Чтобы дополнительно оценить механическую надежность нашего MASC, ориентированного на практические носимые устройства, мы провели измерения GCD при углах изгиба 0 °, 90 °, 180 ° и 360 ° (рис. 5f и вставка). Примечательно, что формы кривых GCD с разными углами изгиба изменяются незначительно, при этом почти 100% начальной емкости сохраняется при изгибе на 360 °, что демонстрирует превосходную механическую гибкость. Такие печатные MASC с их превосходными механическими свойствами, таким образом, способствуют многопольной интеграции с системами солнечных элементов.
Такие печатные MASC с их превосходными механическими свойствами, таким образом, способствуют многопольной интеграции с системами солнечных элементов.
Интеграция гибких автономных устройств с солнечной зарядкой
Быстрое развитие носимой интеллектуальной электроники привело к огромному спросу на гибкие, безопасные и долговечные источники энергии 34 . Напечатанные MASC, как показано выше, могут служить в качестве жизнеспособных устройств хранения энергии, но все же могут не соответствовать требованиям к автономному питанию без внешнего источника зарядки, особенно для носимых приложений. В этом отношении интеграция с компонентом сбора энергии, таким как солнечная батарея, является возможным решением как для захвата, так и для хранения энергии в одном устройстве.Мы начали с оценки нашего гибкого VN // MnO 2 ASC в системе интеграции солнечной зарядки. Для этого в качестве фотоэлектрического модуля использовался недорогой поликристаллический кремниевый солнечный элемент (дополнительный рис. 18a, b), а в качестве модуля накопления энергии использовался квазитвердотельный ASC в собранном виде. На рисунке 6а показаны профили солнечной зарядки / разрядки интегрированного устройства при различной интенсивности света с одинаковой плотностью тока разряда (1 мА · см 90 · 107 −2 90 · 108). Плотность выходного тока солнечного элемента, соответствующая интенсивности света, показана на дополнительном рис.18c. Когда интенсивность света увеличивалась с 4 до 950 Вт / см -2 , плотность выходного тока соответственно увеличивалась с 1,6 до 114 мА / см -2 . Такое резкое увеличение интенсивности света (тока) могло бы значительно сократить продолжительность солнечной зарядки (например, до 3 с). Кроме того, соответственно сокращается время разряда, что можно объяснить увеличением падения ИК-излучения при больших плотностях тока. Стоит отметить, что режим разряда все еще может вернуться к исходному состоянию после набора интенсивности света обратно до 4 Вт · см 90 · 107 −2 90 · 108, что указывает на отличную способность к скорости.
18a, b), а в качестве модуля накопления энергии использовался квазитвердотельный ASC в собранном виде. На рисунке 6а показаны профили солнечной зарядки / разрядки интегрированного устройства при различной интенсивности света с одинаковой плотностью тока разряда (1 мА · см 90 · 107 −2 90 · 108). Плотность выходного тока солнечного элемента, соответствующая интенсивности света, показана на дополнительном рис.18c. Когда интенсивность света увеличивалась с 4 до 950 Вт / см -2 , плотность выходного тока соответственно увеличивалась с 1,6 до 114 мА / см -2 . Такое резкое увеличение интенсивности света (тока) могло бы значительно сократить продолжительность солнечной зарядки (например, до 3 с). Кроме того, соответственно сокращается время разряда, что можно объяснить увеличением падения ИК-излучения при больших плотностях тока. Стоит отметить, что режим разряда все еще может вернуться к исходному состоянию после набора интенсивности света обратно до 4 Вт · см 90 · 107 −2 90 · 108, что указывает на отличную способность к скорости. Другими словами, наш ASC демонстрирует способность выдерживать большие токи (например, 114 мА · см −2 ), превосходя по характеристикам новые аккумуляторы энергии (такие как литий-ионные батареи, LIB 35,36 и конденсаторы, LIC 5 ). Общая эффективность преобразования и хранения энергии ( η всего ) была рассчитана при различной интенсивности света, как показано на рис. 6b. Наивысший η в целом достигается при интенсивности света 4 Вт / см −2 (соответствует типичным условиям освещения в помещении и окружающей среде), достигая замечательного значения 11.95%. Такой высокий общий КПД и отличные характеристики внутреннего освещения можно отнести к высокой стойкости к току и высокой эффективности накопления энергии ( η накопитель ) нашего ASC, где η накопитель рассчитан как ~ 80 %. Чтобы еще больше подтвердить превосходство нашей системы зарядки солнечных батарей, было изготовлено интегрированное устройство GaAs-ASC путем объединения солнечных элементов на основе GaAs с собранными ASC, что продемонстрировало рекордно высокое значение η из 17.
Другими словами, наш ASC демонстрирует способность выдерживать большие токи (например, 114 мА · см −2 ), превосходя по характеристикам новые аккумуляторы энергии (такие как литий-ионные батареи, LIB 35,36 и конденсаторы, LIC 5 ). Общая эффективность преобразования и хранения энергии ( η всего ) была рассчитана при различной интенсивности света, как показано на рис. 6b. Наивысший η в целом достигается при интенсивности света 4 Вт / см −2 (соответствует типичным условиям освещения в помещении и окружающей среде), достигая замечательного значения 11.95%. Такой высокий общий КПД и отличные характеристики внутреннего освещения можно отнести к высокой стойкости к току и высокой эффективности накопления энергии ( η накопитель ) нашего ASC, где η накопитель рассчитан как ~ 80 %. Чтобы еще больше подтвердить превосходство нашей системы зарядки солнечных батарей, было изготовлено интегрированное устройство GaAs-ASC путем объединения солнечных элементов на основе GaAs с собранными ASC, что продемонстрировало рекордно высокое значение η из 17.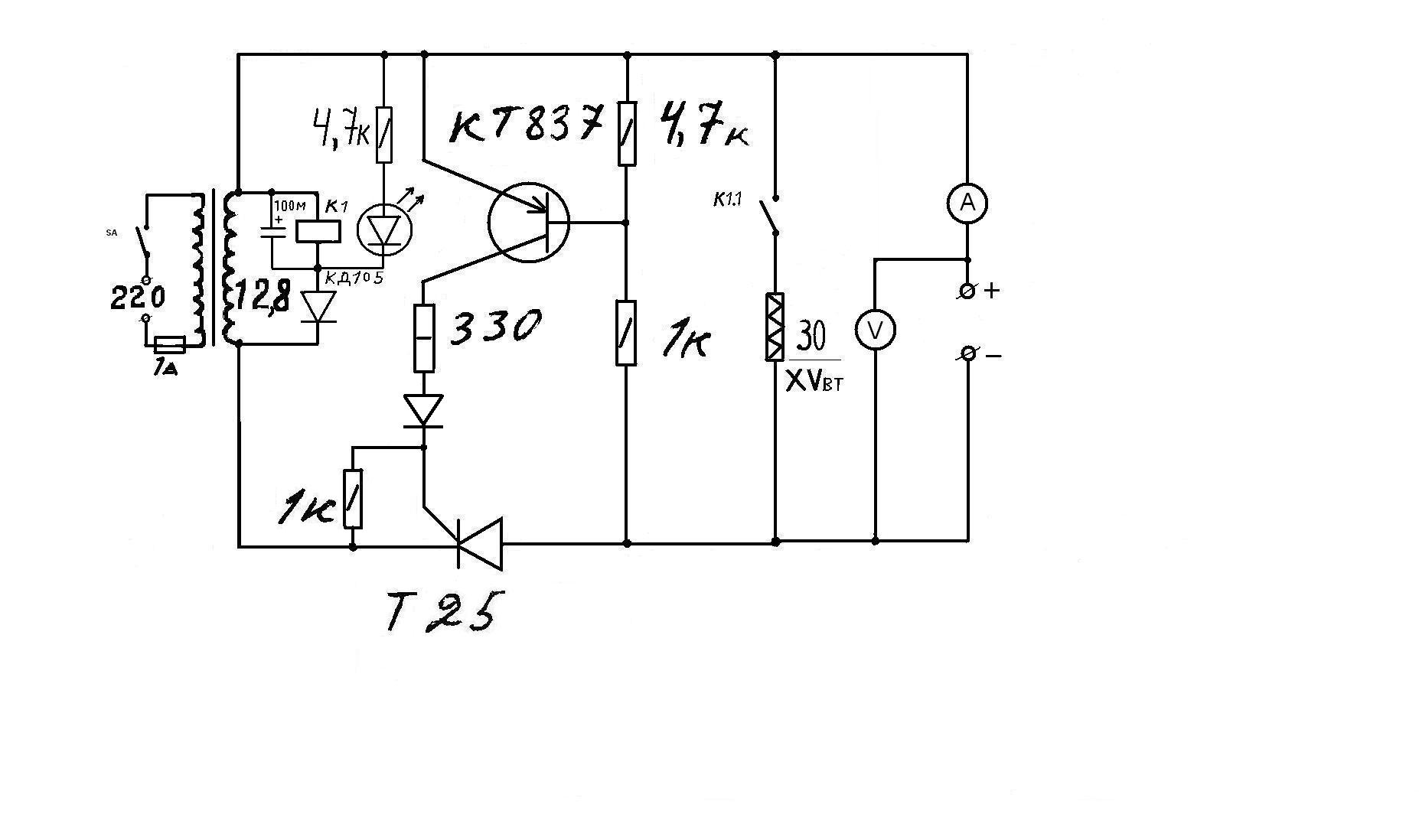 57% при 1000 Вт · м −2 (дополнительный рисунок 19). Такой выдающийся общий КПД можно объяснить высокой эффективностью преобразования солнечного элемента на основе GaAs (25,88%) и высокой эффективностью накопления энергии ASC (67,90%). На рисунке 6c сравниваются η в целом между нашим интегрированным блоком и современными системами зарядки от солнечных батарей. Очевидно, что η в целом наших кремниевых солнечных элементов / ASC и GaAs-солнечных элементов / устройств ASC превосходит другие интегрированные системы, такие как интегрированные в кремниевые солнечные элементы LIB (c-Si-LIB, 7.61%) и перовскитный солнечный элемент в сочетании с LIC (PSC-LIC, 8,41%). Подробное сравнение основных параметров (общий КПД, выходное напряжение и ток выносливости) между текущей работой и другими опубликованными исследованиями в области автономных систем с солнечной зарядкой представлено в дополнительной таблице 4.
57% при 1000 Вт · м −2 (дополнительный рисунок 19). Такой выдающийся общий КПД можно объяснить высокой эффективностью преобразования солнечного элемента на основе GaAs (25,88%) и высокой эффективностью накопления энергии ASC (67,90%). На рисунке 6c сравниваются η в целом между нашим интегрированным блоком и современными системами зарядки от солнечных батарей. Очевидно, что η в целом наших кремниевых солнечных элементов / ASC и GaAs-солнечных элементов / устройств ASC превосходит другие интегрированные системы, такие как интегрированные в кремниевые солнечные элементы LIB (c-Si-LIB, 7.61%) и перовскитный солнечный элемент в сочетании с LIC (PSC-LIC, 8,41%). Подробное сравнение основных параметров (общий КПД, выходное напряжение и ток выносливости) между текущей работой и другими опубликованными исследованиями в области автономных систем с солнечной зарядкой представлено в дополнительной таблице 4.
 6
6 Электрохимический производительность встроенных блоков с солнечной зарядкой. a Кривые солнечной зарядки / разрядки при разной интенсивности света и одинаковой плотности тока разряда 1 мА см −2 . b Вычислено η всего при различных интенсивностях света, соответствующих a . c Сравнение η всего с недавно опубликованной интегрированной энергетической системой с солнечной зарядкой. d Напряжение-время наших гибких интегрированных солнечных батарей. e Кривые заряда / разряда солнечной энергии при разных углах изгиба (левая панель) с цифровыми фотографиями (правая панель), показывающими условия изгиба. f Испытание на долговечность гибкого интегрированного блока с солнечной зарядкой на 100 циклов. г Стабильность при длительной работе гибкого интегрированного блока с солнечной зарядкой. h Демонстрация сценариев носимых устройств с использованием наших интегрированных блоков с солнечной зарядкой
Затем мы рассмотрели практическое использование нашей системы, собрав гибкий интегрированный блок с солнечной зарядкой в соответствии с конфигурацией, изображенной на рис.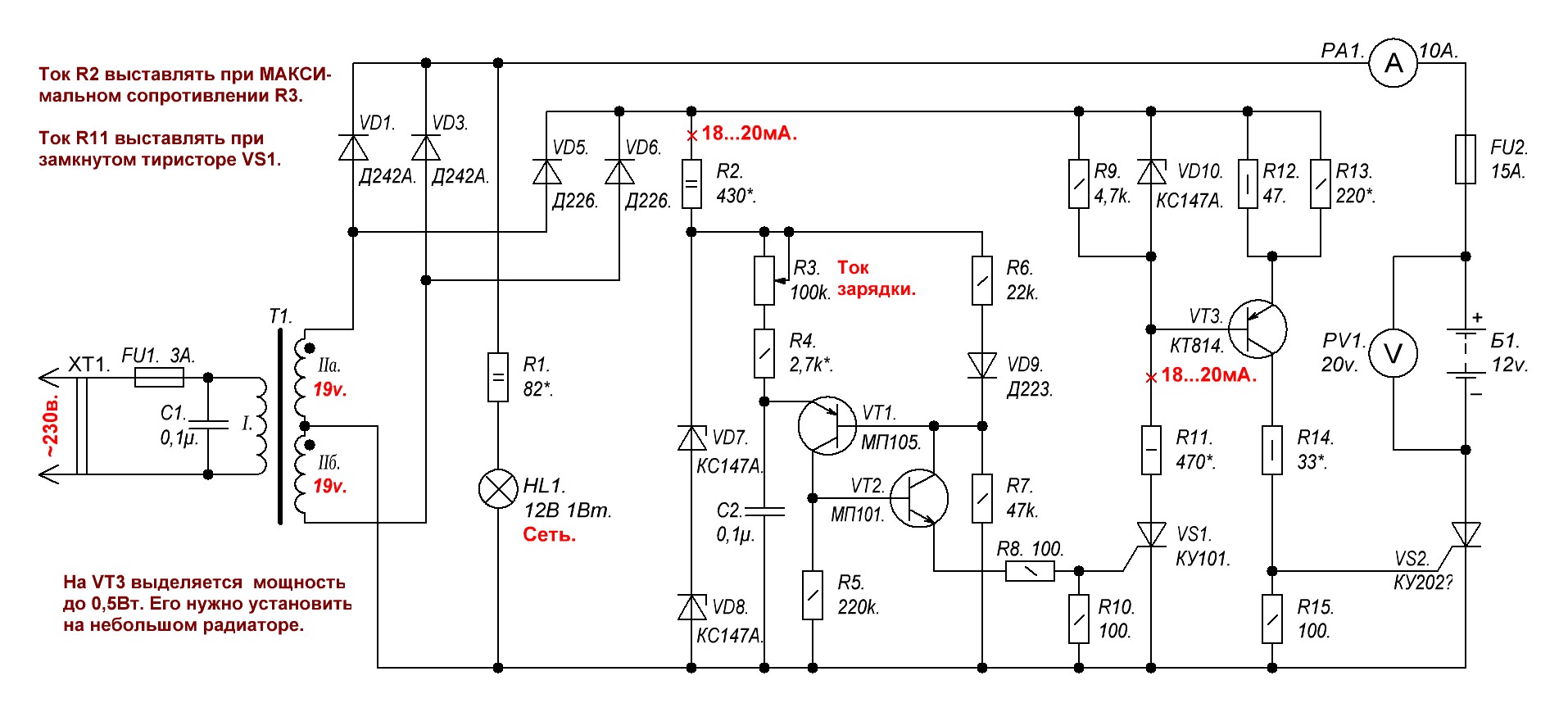 1a. Примечательно, что гибкий солнечный элемент из аморфного кремния функционировал как фотоэлектрический модуль (дополнительный рис.20а, б). VN // MnO 2 MASC был напечатан непосредственно на нижней части солнечного элемента, которая защищена слоем полиимида (PI). На рисунке 6d показаны кривые зарядки / разрядки нашего гибкого интегрированного устройства. При тех же условиях солнечной зарядки 4 Вт / см −2 он показывает хорошие характеристики. η в целом такой гибкой системы достигает 5,2% (дополнительный рисунок 20c). Несмотря на то, что он не такой высокий, как наш жесткий интегрированный блок, он все же превосходит большинство известных гибких фото-зарядных систем с автономным питанием 4,37,38,39,40,41 (см. Дополнительную таблицу 5).Чтобы представить себе реалистичные носимые устройства, мы дополнительно оценили механическую стабильность интегрированных автономных устройств с солнечной зарядкой. На рисунке 6e показаны профили солнечной зарядки (4 Вт · см -2 ) / разрядки (1 мА · см -2 ) при углах изгиба 0 °, 90 ° и 180 ° (как показано на цифровых фотографиях).
1a. Примечательно, что гибкий солнечный элемент из аморфного кремния функционировал как фотоэлектрический модуль (дополнительный рис.20а, б). VN // MnO 2 MASC был напечатан непосредственно на нижней части солнечного элемента, которая защищена слоем полиимида (PI). На рисунке 6d показаны кривые зарядки / разрядки нашего гибкого интегрированного устройства. При тех же условиях солнечной зарядки 4 Вт / см −2 он показывает хорошие характеристики. η в целом такой гибкой системы достигает 5,2% (дополнительный рисунок 20c). Несмотря на то, что он не такой высокий, как наш жесткий интегрированный блок, он все же превосходит большинство известных гибких фото-зарядных систем с автономным питанием 4,37,38,39,40,41 (см. Дополнительную таблицу 5).Чтобы представить себе реалистичные носимые устройства, мы дополнительно оценили механическую стабильность интегрированных автономных устройств с солнечной зарядкой. На рисунке 6e показаны профили солнечной зарядки (4 Вт · см -2 ) / разрядки (1 мА · см -2 ) при углах изгиба 0 °, 90 ° и 180 ° (как показано на цифровых фотографиях). Наблюдение увеличения продолжительности солнечной зарядки в зависимости от изгиба можно в основном объяснить уменьшением эффективной площади освещения изогнутого солнечного элемента.В этом случае энергия разряда, обозначенная временем разряда, остается постоянной, что свидетельствует о хорошей гибкости нашего автономного устройства. Также было проведено испытание на механическую прочность путем многократного изгиба интегрированного устройства на 90 ° в течение 100 циклов (рис. 6f, вставка). Сохранение емкости достигало 94% и 80% после 10 и 100 циклов изгиба соответственно. В ходе дальнейших испытаний были реализованы различные гибкие условия для проверки механической прочности наших устройств (дополнительный рис.21). До и после деформаций складывания, скручивания и волнистости интегрированный блок показал сохранение емкости 97,50%, 85,22% и 93,04% соответственно. Все это свидетельствует о хорошей гибкости нашего автономного блока для практических применений.
Наблюдение увеличения продолжительности солнечной зарядки в зависимости от изгиба можно в основном объяснить уменьшением эффективной площади освещения изогнутого солнечного элемента.В этом случае энергия разряда, обозначенная временем разряда, остается постоянной, что свидетельствует о хорошей гибкости нашего автономного устройства. Также было проведено испытание на механическую прочность путем многократного изгиба интегрированного устройства на 90 ° в течение 100 циклов (рис. 6f, вставка). Сохранение емкости достигало 94% и 80% после 10 и 100 циклов изгиба соответственно. В ходе дальнейших испытаний были реализованы различные гибкие условия для проверки механической прочности наших устройств (дополнительный рис.21). До и после деформаций складывания, скручивания и волнистости интегрированный блок показал сохранение емкости 97,50%, 85,22% и 93,04% соответственно. Все это свидетельствует о хорошей гибкости нашего автономного блока для практических применений.
На рисунке 6g также представлены характеристики циклического режима фото-зарядки / разрядки, испытанные при интенсивности света 4 Вт / см −2 и плотности тока разряда 1 мА · см −2 , причем на вставке показаны первые 10 циклов и последние 10 циклов кривых фото-зарядки / разрядки. Примечательно, что наш интегрированный блок сохранил выдающуюся стабильность при циклических нагрузках с удивительно высоким сохранением емкости 98,7% после 100 циклов, что демонстрирует превосходную надежность и механическую стабильность. Ожидается, что наши интегрированные автономные блоки с солнечной зарядкой, обладающие хорошей гибкостью, длительным сроком службы и высокой безопасностью, будут жизнеспособными для многих сценариев использования носимых устройств. На рисунке 6h показана демонстрация концепции. Интегрированный блок можно полностью зарядить при воздействии естественного солнечного света или наружного освещения (т.е., в дневное время). Гибкость и даже возможность сворачивания такого блока питания позволяет носить его прямо на одежде или наклеивать на сумки в качестве эффективного автономного источника энергии. Энергия, хранящаяся в заряжаемом солнечной батареей MASC, может в конечном итоге использоваться для питания светодиодной панели, особенно для использования внутри помещений в ночное время.
Примечательно, что наш интегрированный блок сохранил выдающуюся стабильность при циклических нагрузках с удивительно высоким сохранением емкости 98,7% после 100 циклов, что демонстрирует превосходную надежность и механическую стабильность. Ожидается, что наши интегрированные автономные блоки с солнечной зарядкой, обладающие хорошей гибкостью, длительным сроком службы и высокой безопасностью, будут жизнеспособными для многих сценариев использования носимых устройств. На рисунке 6h показана демонстрация концепции. Интегрированный блок можно полностью зарядить при воздействии естественного солнечного света или наружного освещения (т.е., в дневное время). Гибкость и даже возможность сворачивания такого блока питания позволяет носить его прямо на одежде или наклеивать на сумки в качестве эффективного автономного источника энергии. Энергия, хранящаяся в заряжаемом солнечной батареей MASC, может в конечном итоге использоваться для питания светодиодной панели, особенно для использования внутри помещений в ночное время.
Дальнейшие исследования происхождения асимметричного разделения заряда в гомодимерах белка
J Am Soc Mass Spectrom. Авторская рукопись; доступно в PMC 2006 6 января.
Опубликован в окончательной редакции как:
PMCID: PMC1325214
NIHMSID: NIHMS3986
Департамент химии Калифорнийского университета в Беркли, Беркли, Калифорния, США Химия, Калифорнийский университет в Беркли, Беркли, Калифорния 94720, США. Электронное письмо: ude.yelekreb.mehcc@smailliw См. другие статьи в PMC, в которых цитируется опубликованная статья.
Abstract
Диссоциация протонированных в газовой фазе белковых димеров на составляющие их мономеры может приводить к симметричному или асимметричному распределению заряда.Диссоциация гомодимеров α -лактальбумина с 15+ зарядами приводит к симметричному, но широкому распределению белковых мономеров с зарядовыми состояниями, сосредоточенными вокруг 8 + / 7 +. Напротив, диссоциация гетеродимера 15+, состоящего из одной молекулы в окисленной форме и одной в восстановленной форме, приводит к сильно асимметричному распределению зарядов, при котором восстановленные частицы уносят преимущественно заряды 11+, а окисленная молекула уносит заряды 4+. . Этот результат не может быть адекватно объяснен дифференциальной зарядкой, происходящей ни в растворе, ни в процессе электрораспыления, но, по-видимому, лучше всего объясняется восстановленными частицами, разворачивающимися при активации в газовой фазе с последующим разделением и переносом протонов на разворачивающиеся частицы в диссоциативном комплексе. чтобы минимизировать кулоновское отталкивание.Для димеров цитохрома c , образованных непосредственно из раствора, зарядовое состояние 17+ подвергается симметричному распределению заряда, тогда как диссоциация 13+ асимметрична. Уменьшение зарядового состояния димеров с зарядами 17+ до 13+ посредством переноса протона в газовой фазе и последующей диссоциации выбранных по массе ионов 13+ приводит к симметричному распределению заряда. Этот результат ясно показывает, что структура димерных ионов с зарядами 13+ зависит от метода образования иона и что структурные различия ответственны за наблюдаемое симметричное и асимметричное разделение зарядов.
. Этот результат не может быть адекватно объяснен дифференциальной зарядкой, происходящей ни в растворе, ни в процессе электрораспыления, но, по-видимому, лучше всего объясняется восстановленными частицами, разворачивающимися при активации в газовой фазе с последующим разделением и переносом протонов на разворачивающиеся частицы в диссоциативном комплексе. чтобы минимизировать кулоновское отталкивание.Для димеров цитохрома c , образованных непосредственно из раствора, зарядовое состояние 17+ подвергается симметричному распределению заряда, тогда как диссоциация 13+ асимметрична. Уменьшение зарядового состояния димеров с зарядами 17+ до 13+ посредством переноса протона в газовой фазе и последующей диссоциации выбранных по массе ионов 13+ приводит к симметричному распределению заряда. Этот результат ясно показывает, что структура димерных ионов с зарядами 13+ зависит от метода образования иона и что структурные различия ответственны за наблюдаемое симметричное и асимметричное разделение зарядов. Это указывает на то, что асимметрия, наблюдаемая, когда эти ионы образуются непосредственно из раствора, должна происходить либо из-за различий в конформациях мономера в димере, которые существуют в растворе, либо в процессе ионизации электрораспылением. Эти результаты предоставляют дополнительные доказательства происхождения асимметрии заряда, которая возникает при диссоциации многозарядных белковых комплексов, и показывают, что некоторая информация о фазе раствора может быть получена из этих экспериментов по газофазной диссоциации.
Это указывает на то, что асимметрия, наблюдаемая, когда эти ионы образуются непосредственно из раствора, должна происходить либо из-за различий в конформациях мономера в димере, которые существуют в растворе, либо в процессе ионизации электрораспылением. Эти результаты предоставляют дополнительные доказательства происхождения асимметрии заряда, которая возникает при диссоциации многозарядных белковых комплексов, и показывают, что некоторая информация о фазе раствора может быть получена из этих экспериментов по газофазной диссоциации.
Биологическая функциональность белков и белковых комплексов сильно зависит от их структур в фазе раствора. Важность области структурной биологии отражена в нескольких недавно присужденных Нобелевских премиях за вклад в методы структурного выяснения, включая ЯМР (Курт Вутрих, 2002 г. по химии), рентгеновскую кристаллографию (Иоганн Дайзенхофер, Роберт Хубер и Хартмут Мишель, 1988 г. в. химия; Герберт Хауптман и Джером Карл 1985 по химии) и кристаллографическая электронная микроскопия (сэр Аарон Клаг 1982 по химии). Анализ белков и белковых комплексов этими методами может предоставить подробную трехмерную информацию, но эти методы часто ограничены экспериментальными ограничениями, такими как необходимость получения полезных кристаллов белка или требование значительного количества образца. Напротив, для масс-спектрометрии требуется небольшой образец, а с ионизацией электрораспылением (ESI) или мягкой лазерной десорбцией / ионизацией (John Fenn and Kiochi Tanaka, 2002 по химии) в масс-спектрометр можно ввести большое количество биомолекул и специфических комплексов.Был разработан ряд методов для исследования общих трехмерных форм ионов биомолекул и нековалентных комплексов с использованием методов ионной подвижности [1–8], H / D обмена как в растворе [9, 10], так и в газовой фазе [ 11–28] и реакционной способности с переносом протона [29, 30].
Анализ белков и белковых комплексов этими методами может предоставить подробную трехмерную информацию, но эти методы часто ограничены экспериментальными ограничениями, такими как необходимость получения полезных кристаллов белка или требование значительного количества образца. Напротив, для масс-спектрометрии требуется небольшой образец, а с ионизацией электрораспылением (ESI) или мягкой лазерной десорбцией / ионизацией (John Fenn and Kiochi Tanaka, 2002 по химии) в масс-спектрометр можно ввести большое количество биомолекул и специфических комплексов.Был разработан ряд методов для исследования общих трехмерных форм ионов биомолекул и нековалентных комплексов с использованием методов ионной подвижности [1–8], H / D обмена как в растворе [9, 10], так и в газовой фазе [ 11–28] и реакционной способности с переносом протона [29, 30].
Для анализа нековалентных комплексов масс-спектрометрия имеет то преимущество, что стехиометрию комплекса можно определить с помощью простого измерения массы [31]. В некоторых случаях MS может обеспечить правильные константы связывания в фазе раствора для белок-лигандных взаимодействий [32–35].Эти разработки подробно рассмотрены [31, 35]. В принципе, информацию о структуре нековалентных комплексов можно потенциально получить, исследуя ионы продуктов, образующиеся в результате диссоциации комплекса, в эксперименте тандемной масс-спектрометрии (МС / МС). Объем соответствующей информации, которую можно получить в результате этих экспериментов, зависит от количества структурной информации о фазе раствора, которая сохраняется биологическими комплексами при переходе в газовую фазу (или от того, может ли эта информация быть восстановлена на основе сведений о происходящих структурных изменениях. во время этого перехода) и степень, в которой методы MS / MS нарушают любую оставшуюся структурную информацию.Многие эксперименты показывают, что газофазные комплексы биомолекул и даже некоторые небольшие молекулы могут сохранять «память» о своей фазовой структуре раствора, и что эта память может быть исследована процессами диссоциации [36–48].
В некоторых случаях MS может обеспечить правильные константы связывания в фазе раствора для белок-лигандных взаимодействий [32–35].Эти разработки подробно рассмотрены [31, 35]. В принципе, информацию о структуре нековалентных комплексов можно потенциально получить, исследуя ионы продуктов, образующиеся в результате диссоциации комплекса, в эксперименте тандемной масс-спектрометрии (МС / МС). Объем соответствующей информации, которую можно получить в результате этих экспериментов, зависит от количества структурной информации о фазе раствора, которая сохраняется биологическими комплексами при переходе в газовую фазу (или от того, может ли эта информация быть восстановлена на основе сведений о происходящих структурных изменениях. во время этого перехода) и степень, в которой методы MS / MS нарушают любую оставшуюся структурную информацию.Многие эксперименты показывают, что газофазные комплексы биомолекул и даже некоторые небольшие молекулы могут сохранять «память» о своей фазовой структуре раствора, и что эта память может быть исследована процессами диссоциации [36–48].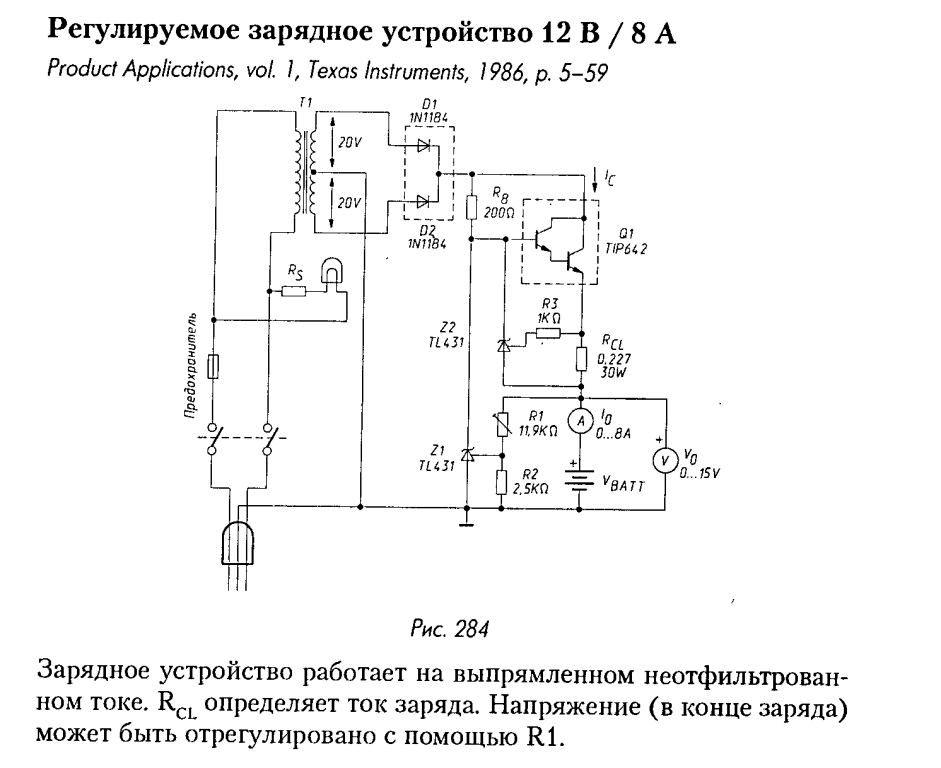
Ряд исследований гомогенных биологических комплексов (тетрамеры, пентамеры, гексамеры и т. Д.) С помощью МС / МС выявили первоначально неожиданный результат, заключающийся в том, что первичный путь диссоциации мультимерных комплексов представляет собой выброс высоко заряженного мономера, который удаляет 30-50 % заряда комплекса [49–53].Рентгеновская кристаллография показывает, что структуры растворов субъединиц во многих гомогенных комплексах идентичны. В некоторых случаях весь комплекс кристаллизуется внутри асимметричной единицы кристалла, как в случае с шига-подобным токсином [9], в то время как в других, таких как апострептавидин, асимметричная единица содержит одну субъединицу, и структура комплекс экстраполирован с помощью молекулярного моделирования [54]. Из-за практически идентичных структур фазы раствора участвующих субъединиц в этих биологических комплексах наблюдаемое асимметричное распределение заряда, по-видимому, является артефактом процедуры ESI-MS / MS.Было высказано предположение, что асимметричное распределение заряда коррелирует с газофазными конформационными перестройками [50, 52, 55, 56]. Также было продемонстрировано, что степень асимметричного распределения заряда для гомодимеров белков зависит от конформационной гибкости белков в комплексе, зарядового состояния комплекса, отложения внутренней энергии и состава растворов для электрораспыления, из которых комплексы образуются. формируются [56]. Для более крупных кластеров, состоящих из идентичных субъединиц, наблюдалось деление с преимущественным образованием протонированных димеров, а с увеличением зарядового состояния и размера кластера наблюдалось усиление образования протонированного мономера [57].Степень деления по сравнению с потерями нейтралов для этих кластеров может быть связана с наблюдаемой для многозарядных металлических кластеров с одним важным отличием: различия в конформационной гибкости субъединиц мономера внутри кластера приводят к различиям в коэффициентах ветвления различных диссоциаций. процессы, наблюдаемые для этих многократно протонированных кластеров биомолекул [57].
Также было продемонстрировано, что степень асимметричного распределения заряда для гомодимеров белков зависит от конформационной гибкости белков в комплексе, зарядового состояния комплекса, отложения внутренней энергии и состава растворов для электрораспыления, из которых комплексы образуются. формируются [56]. Для более крупных кластеров, состоящих из идентичных субъединиц, наблюдалось деление с преимущественным образованием протонированных димеров, а с увеличением зарядового состояния и размера кластера наблюдалось усиление образования протонированного мономера [57].Степень деления по сравнению с потерями нейтралов для этих кластеров может быть связана с наблюдаемой для многозарядных металлических кластеров с одним важным отличием: различия в конформационной гибкости субъединиц мономера внутри кластера приводят к различиям в коэффициентах ветвления различных диссоциаций. процессы, наблюдаемые для этих многократно протонированных кластеров биомолекул [57].
Газофазный водород-дейтериевый (H / D) обмен обычно осуществляется путем хранения интересующего иона в вакууме и измерения изменения массы иона из-за реакций обмена H / D, которые происходят во время столкновений с нейтральными молекулами обменного реагента. , например, D 2 O или ND 3 .Хотя газофазный обмен H / D широко использовался для характеристики пептидов и малых молекул [11–21], а также структуры некоторых газофазных белков [22–24], очень мало исследований было выполнено с нековалентными комплексами, включающими биологические ионы, и эта нековалентная работа была сосредоточена на димерах пептидов [25, 26] и пептидных комплексах с ионами металлов [16, 18, 21, 27] и небольшими молекулами [15, 28].
, например, D 2 O или ND 3 .Хотя газофазный обмен H / D широко использовался для характеристики пептидов и малых молекул [11–21], а также структуры некоторых газофазных белков [22–24], очень мало исследований было выполнено с нековалентными комплексами, включающими биологические ионы, и эта нековалентная работа была сосредоточена на димерах пептидов [25, 26] и пептидных комплексах с ионами металлов [16, 18, 21, 27] и небольшими молекулами [15, 28].
Здесь представлена дальнейшая характеристика гомодимеров белка с использованием газофазной диссоциации и газофазного H / D обмена.Исследования диссоциации смешанных димеров окисленного и восстановленного α -лактальбумина предоставляют дополнительные доказательства того, что конформационная гибкость играет ключевую роль в определении степени асимметричного распределения заряда. Для диссоциации гомодимеров цитохрома c показано, что зарядовые состояния, полученные в результате снижения заряда в газовой фазе из гомодимеров с более высоким зарядовым состоянием, диссоциируют совсем иначе, чем состояния, полученные непосредственно из раствора. Представлена первая демонстрация газофазного H / D обмена белкового комплекса.Эти результаты показывают, что два разных механизма ответственны за асимметричное распределение заряда, наблюдаемое при диссоциации гомодимеров белка.
Experimental
Эксперименты проводились на масс-спектрометре Berkeley-Bruker с узким проходом 9,4 Тл с ионно-циклотронным резонансом с преобразованием Фурье (FT-ICR), который описан в другом месте [56]. Используемые экспериментальные процедуры аналогичны использованным ранее [56]. Краткое описание и дополнительные сведения приведены ниже.
Chemicals
Цитохром сердца лошади c , α -лактальбумин из коровьего молока и триэтиламин были приобретены у Sigma-Aldrich Co. (Сент-Луис, Миссури) и использовались без дополнительной очистки. Дейтерированный аммиак был приобретен в Cambridge Isotope Laboratories (Woburn, MA). Ацетамидирование α -лактальбумина было выполнено доктором Дэвидом Кингом (Калифорнийский университет, Беркли). Все четыре внутримолекулярные дисульфидные связи α -лактальбумина восстанавливаются трис (карбоксиэтил) фосфином, ацетамидируются в темноте йодацетамидом в течение одного часа и очищаются с помощью ВЭЖХ для удаления буфера и непрореагировавших реагентов.Практически полное восстановление и ацетамидирование всех дисульфидных связей в α -лактальбумине было подтверждено измерениями массы.
Образование димеров
Образование неспецифических белковых димеров усиливается за счет использования концентраций белка 100–500 мкМ M и регулирования напряжений на границе раздела для «мягкого» введения ионов. Сигнал димера улучшается за счет настройки на селективное накопление ионов во внешнем гексаполе в области источника прибора. Это достигается накоплением ионов в гексаполе в течение 3–4 с (против ≤ 1.0 с), увеличивая смещение постоянного тока гексаполя с ~ 2,7 В до 3,5–4,5 В и вводя несколько скоплений гексаполя в ионную ячейку перед обнаружением.
Смешанные димеры окисленного (все четыре дисульфидные связи не повреждены) и восстановленного (все четыре дисульфидные связи разорваны и все восемь остатков цистеина впоследствии ацетамидированы) α -лактальбумин были образованы путем смешивания растворов двух форм α -лактальбумина в различных соотношения. Концентрация общего белка поддерживалась постоянной на уровне ~ 200 мкМ М, и отношения двух компонентов были изменены, чтобы максимизировать абсолютное содержание смешанного димера.Наибольшее количество смешанного димера появляется при относительной концентрации восстановленного до окисленного α -лактальбумина ~ 2: 1. Поскольку концентрации белка определялись массой лиофилизированных порошков, используемых для приготовления раствора, эти концентрации являются приблизительными. Ионы димера белка формировали с использованием ESI со скоростью потока ~ 1 мкМ л / мин в условиях раствора 1: 1 вода: метанол + 2% уксусная кислота.
Димеры цитохрома c были сформированы с использованием обычного ESI при скорости потока 1-2 мкМ л / мин и концентрации 100 мкМ М в 1: 1 вода: метанол + 2% уксусная кислота.Зарядовые состояния димеров, сформированные непосредственно из раствора, варьируются от [(цитохром c ) 2 + 19H] 19+ до [(циктохром c ) 2 + 13H] 13+ (сокращенно D19 – D13) . Зарядовые состояния димера с m / z выше, чем у D17, были выброшены из ионной ячейки с использованием коррелированных выстрелов, оставляя зарядовые состояния D19 – D17, оставшиеся в ионной ячейке. Вакуумная камера FT-ICR была подготовлена для снижения заряда путем кратковременного введения триэтиламина в вакуумную камеру под давлением ~ 3 × 10 90 · 10 7 -8 90 · 108 торр с использованием пьезоэлектрического импульсного клапана.Импульсный клапан затем закрывался через 5 с, и базовое давление прибора возвращалось к ~ 8 × 10 90 · 107 -9 90 · 108 торр через 95 с. Снижение заряда посредством переноса протона осуществлялось путем улавливания гомодимеров цитохрома c с зарядовыми состояниями D19 – D17 в приборе до тех пор, пока столкновения с остаточными молекулами триэтиламина не снижали зарядовое состояние димеров в течение 100 с, так что 13+ было самая распространенная форма димера. Вновь образованные ионы D13 были изолированы с использованием коррелированных свип-сигналов и диссоциированы с помощью длительной диссоциации, активируемой столкновениями (SORI-CAD) [58] вне резонанса и вне резонанса, при +600 Гц вне резонанса и 3.5 В пик – пик в течение 0,25 с после подачи импульса азота, повышающего давление в вакуумной камере до ~ 10 –6 торр.
Газофазный H / D обмен белковых комплексов
Газофазный H / D обмен гомодимеров цитохрома c осуществляли путем улавливания всех генерируемых электрораспылением ионов в ячейке и введения ND 3 в вакуумную камеру с использованием ручной герметичный клапан (Varian, Lexington, MA) на 180 с при нескольких давлениях в диапазоне от ~ 1 × 10 −8 до максимального ~ 3 × 10 −7 торр.После 180 с замены клапан утечки был закрыт, и вакуумная камера откачивалась в течение 60–120 с, пока базовое давление не стало достаточно низким для изотопного разрешения (<1 × 10 –8 торр). Предыдущие эксперименты по газофазному обмену H / D с белками использовали D 2 O в качестве обменного реагента [22–24, 59]. Из-за трудностей, связанных с длительным временем улавливания (> 300 с) и значительным временем, необходимым для удаления относительно нелетучего D 2 O, в качестве обменного реагента был выбран ND 3 .Давление вводимого в вакуумную камеру НД 3 измерялось некалиброванным ионным манометром, расположенным на расстоянии ~ 1,1 м от ионной ячейки.
Результаты и обсуждение
Диссоциация смешанных
димеров α -лактальбуминаИспользуя растворы ESI с высокими концентрациями белка, можно образовать как гомодимеры окисленного белка α -лактальбумина (каждый белок имеет четыре интактных дисульфидных связи), так и окисленно-восстановленные гетеродимеры α -лактальбумина (один белок имеет четыре интактных дисульфидных связи, а другой – четыре восстановленные дисульфидные связи, причем все восемь остатков цистеина ацетамидированы).И окисленные гомодимеры, и окисленно-восстановленные гетеродимеры образуются непосредственно из ESI с 15 присоединенными протонами ((Ox-Ox) 15 и (Ox-Red) 15, соответственно) (). (Ox-Ox) 15 диссоциирует с довольно широким, но симметричным распределением заряда (), аналогичным тому, что наблюдалось ранее для (Ox-Ox) 13 и (Red-Red) 17 [56]. (Red-Red) 13, как было ранее показано, асимметрично диссоциирует с преимущественным образованием M9 / M4 [56]. В отличие от (Ox-Ox) 15, (Ox-Red) 15 диссоциирует с сильно асимметричным распределением заряда ().Наиболее распространенным процессом диссоциации димера является (Ox-Red) 15 → Red11 + Ox4, процесс разделения, при котором восстановленный α -лактальбумин удаляет почти всего заряда! Обратите внимание, что большая интенсивность сигнала Red11 по сравнению с Ox4 обусловлена как более низким уровнем заряда последнего (сигнал FT-ICR пропорционален заряду), так и несколько меньшей эффективностью обнаружения при более высоком m / z .
( a ) Масс-спектр ESI, полученный из 200 мкМ раствора M (общая концентрация белка), содержащего окисленный (нетронутый дисульфид) и восстановленный α -лактальбумин (1: 1 вода: метанол + 2% уксусная кислота) , ( b ) МС / МС спектр окисленного-восстановленного гетеродимера α -лактальбумина с 15 протонами, (Ox-Red) 15 и ( c ) МС / МС спектр окисленного α -лактальбумина -окисленный гомодимер (Ox-Ox) 15, образованный из раствора ~ 125 мкМ М окисленного α -лактальбумина в 1: 1 вода: метанол + 2% уксусная кислота.Red8 представляет (восстановленный α -лактальбумин + 8H) 8+ и т. Д.
Происхождение сильно асимметричного распределения заряда может быть связано с асимметричным разделением, которое происходит в газовой фазе [56], или с асимметрией конформации или заряд мономеров, составляющих димер, либо в растворе, либо индуцированный процессами электрораспыления. В первом механизме асимметричное распределение заряда происходит из-за конформационного изменения одной из субъединиц мономера, которая составляет димер, после активации.Когда мономер разворачивается, молекула преодолевает активационный барьер для разворачивания и разделения. Из-за кулоновской энергии выход из этого барьера является сильно отталкивающим, и когда одна часть разворачивающейся молекулы выходит из этого канала, она будет управлять разворачиванием остальной части молекулы, когда она отделяется от другой субъединицы мономера. В этом процессе протоны будут переходить к выходящему разворачивающемуся иону, чтобы минимизировать кулоновскую энергию [30]. Поскольку восстановленный белок более гибок конформационно и может разворачиваться в значительно большей степени, чем окисленный белок, восстановленный белок разворачивается и уносит больший заряд, чем окисленная форма.
Альтернативный механизм этой асимметрии заряда заключается в том, что (Ox-Red) 15 может образовываться в растворе путем агрегации сильно заряженных восстановленных и низкозарядных окисленных мономеров α -лактальбумина в растворе или образовываться во время электрораспыления за счет асимметричной зарядки. отдельных белковых составляющих на заключительных этапах ESI [60]. Также было высказано предположение, что асимметричное распределение заряда для гетеродимеров может быть связано с различиями в pI [61, 62]. Хотя возникает разница в зарядке этих двух молекул, она не учитывает величину асимметричного разделения заряда.В масс-спектре ESI смешанного раствора α -лактальбумина восстановленный α -лактальбумин появляется преимущественно при 8+ и в меньшей степени при 9+ и 10+ (). Наблюдается очень мало 11+. Окисленный α -лактальбумин встречается только в зарядовом состоянии 8+. В спектре диссоциации (Ox-Red) 15 доминирующими наблюдаемыми видами являются Red11 и Ox5, несмотря на то, что численность этих видов, продуцируемых непосредственно ESI, незначительна. Хотя можно было бы рационализировать более низкое зарядовое состояние, наблюдаемое для Ox, как немного более низкое зарядовое состояние димера по сравнению с суммой зарядовых состояний отдельных частиц, это не может быть верным для восстановленных частиц, которые имеют значительно более высокий заряд при диссоциации. спектра, чем при формировании непосредственно электрораспылением.Таким образом, этот результат трудно рационализировать, основываясь только на модели, в которой асимметричное распределение заряда является либо реликтом агрегации фазы раствора, либо артефактом процесса электрораспыления. Результаты гетеродимеров (Ox-Red) 15 α -лактальбумина, по-видимому, лучше всего подтверждают процесс асимметричного распределения заряда в газовой фазе [56].
Диссоциация димеров цитохрома c
Гомодимеры цитохрома c легко образуются из растворов для электрораспыления высокой концентрации (~ 100 мкм M) с зарядовыми состояниями в диапазоне от [(цитохром c ) 2 + 19H] 19 + к [(цитохром c ) 2 + 13H] 13+ (сокращенно D19 – D13).Как было показано ранее, диссоциация цитохрома c D19, образованного непосредственно из раствора, приводит к узкому симметричному процессу разделения заряда [56]. МС / МС D17, образованного непосредственно из раствора, приводит к относительно широкому, но симметричному процессу разделения заряда (преимущественно D17 → M9 + M8) ().
МС / МС спектры ( a ) (димеры цитохрома c + 17H) 17+ , (D17), образованные непосредственно ESI, ( b ) (димеры цитохрома c + 13H) 13+ , (D13), образованный выделением D19-D17, восстановлением газофазного переноса протона диэтиламином, выделением и последующей активацией D13, и ионы ( c ) D13, образованные непосредственно из раствора.Ионы образовывались из 100 мМ раствора (вода: метанол, 1: 1 + 2% уксусная кислота).
Напротив, диссоциация D13, образованного непосредственно из раствора, приводит к сильно асимметричному распределению заряда с центром вокруг D13 → M9 + M4 (). Ионы D17-D19 были изолированы и прореагировали с триэтиламином внутри ионной ячейки FT-ICR в течение 100 с, что привело к образованию преимущественно ионов D13. Выделение D13 и последующая активация этих ионов дает спектр МС / МС, показанный на. Доминирующим процессом является симметричная диссоциация с центром D13 → M7 + M6.Таким образом, процесс диссоциации D13 явно зависит от того, образуются ли они в результате восстановления с переносом протона в газовой фазе D17 – D19 () или образуются непосредственно из раствора (). В этих экспериментальных условиях внутренняя энергия иона уравновешивается с полем излучения черного тела при комнатной температуре до активации иона [63]. Таким образом, различие во фрагментации не может быть артефактом различия внутренней энергии. Эти результаты показывают, что структуры ионов D13 различаются для этих двух методов образования ионов и что структура димера влияет на то, как эти димеры диссоциируют, т.е.е., через симметричное и асимметричное разделение заряда. Следует отметить, что димеризация в растворе неспецифична и что может присутствовать большой ансамбль димерных структур. Однако ясно, что существуют по крайней мере две отдельные структуры или различные ансамбли структур.
«Память» ионов D13 может быть связана с различными димерными структурами, существующими в растворе. Эти димеры могут получать разное количество зарядов в процессе ионизации электрораспылением, или процесс электрораспыления может вызывать конформационные изменения в некоторой части димеров при переходе из раствора в газовую фазу, что приводит к различным структурам димеров для D17 по сравнению с D13.Мы не можем четко различать эти две возможности, основываясь только на этих результатах. Однако широкое распределение в зарядовых состояниях димеров, образованных в этих денатурирующих условиях, по сравнению с типичным единственным доминирующим зарядовым состоянием димера, наблюдаемым при электрораспылении из растворов, в которых эти молекулы имеют нативную структуру, предполагает, что первый механизм более вероятен, т. Е. в растворе существуют по крайней мере две отдельные структуры или ансамбль структур, и эти структуры в конечном итоге имеют разную степень заряда в процессе электрораспыления.
Аналогичные результаты наблюдаются для ионов D15, образованных из раствора, по сравнению с ионами, образованными переносом протона. Те, которые образуются непосредственно из раствора, демонстрируют бимодальную диссоциацию: симметричный процесс, сосредоточенный вокруг M8 / M7, и асимметричный процесс, сосредоточенный вокруг M10 / M5 (). Восстановление заряда D19 – D17 с образованием более низких зарядовых состояний с последующим выделением и диссоциацией D15 приводит только к симметричной диссоциации с образованием M8 / M7 (). Опять же, эти ионы D15 явно сохраняют «память» о том, образовались ли они в газовой фазе из более высоких зарядовых состояний, которые симметрично диссоциируют, или непосредственно из раствора.
МС / МС спектры (цитохрома c димеры + 15H) 15+ , (D15), образованного ( a ) непосредственно ESI и ( b ), образованного выделением D19-D17, газ- восстановление фазового переноса протона диэтиламином, выделение и последующая активация D15. Ионы образовывались из 100 мМ раствора (вода: метанол, 1: 1 + 2% уксусная кислота).
Газофазный H / D обмен мономеров и димеров цитохрома c с ND
3Предыдущие исследования показали, что степень асимметрии заряда цитохрома c D11, образованного непосредственно наноэлектрораспылением с использованием водных растворов, содержащих 100 мМ ацетата аммония нейтральный pH зависел от внутренней энергии, которая вкладывалась в ионы [56].Чтобы попытаться установить, в какой степени различные конформеры могут вносить вклад в наблюдаемую энергетическую зависимость, на D11 были проведены исследования обмена H / D. Тщательная настройка параметров источника ESI, в частности увеличение напряжения смещения на внешнем гексаполе накопления и увеличение времени нагрузки гексаполя, способствует образованию цитохрома c D11 ().
ESI масс-спектр 500 мкМ M конский цитохром c в водном растворе со 100 мМ ацетатом аммония, pH ~ 7, со спектрометром, настроенным на получение максимального количества димеров с зарядами 11+ (D11).
Для этих экспериментов по газофазному H / D обмену давление NH 3 изменялось от 1 × 10 –8 до 3 × 10 –7 торр с постоянным временем реакции 180 с. Для эксперимента при самом высоком давлении реагента (наибольшая степень обмена H / D) () среднее количество атомов водорода, обмениваемых на дейтерий, составляет ~ 170 после вычитания обмена 11 протонов. Ширина распределения составляет примерно 110 Да. Отношение сигнал / шум недостаточно, чтобы приписать вариации интенсивностей изотопов разрешимым конформерам.Напротив, M7 в тех же условиях показывает средний обмен ~ 84 атомов водорода на дейтерий с шириной изотопического распределения ~ 50. Таким образом, как средняя степень обмена, так и ширина распределения димера примерно вдвое больше, чем у мономера. Если бы присутствовала только одна структура, нельзя было бы ожидать, что суммарная максимальная степень обмена и ширина распределения будут линейно увеличиваться с массой иона, если предположить, что степень обмена H / D связана с открытой поверхностью кластера.Эти результаты предполагают присутствие множественных конформеров димеров, которые не разрешаются в этих экспериментах по обмену H / D, что согласуется с нашим ожиданием, что эти ионы D11 образуются в результате неспецифической димеризации молекул циктохрома c в растворе.
Частичные масс-спектры ESI цитохрома c (показана область D11) при тех же условиях раствора, что и раньше, и после газофазного водородно-дейтериевого обмена с ND 3 в течение 180 с при ( b ) 1 × 10 −7 торр и ( c ) 3 × 10 −7 торр.
Обширные исследования газофазного H / D обмена мономеров цитохрома c были проведены McLafferty и соавторами [22–24], которые обнаружили целых семь конформаций цитохрома c M7, образованных непосредственно ESI [24]. В наших экспериментах в качестве обменного реагента используется ND 3 вместо D 2 O [24], и время реакции короче. Распределение изотопов для M7 предполагает наличие нескольких конформеров, согласующихся с результатами, полученными Маклафферти и соавторами.
Выводы
Асимметричное распределение заряда, которое наблюдается при диссоциации гомодимеров белков, можно объяснить двумя различными механизмами. В одном механизме асимметрия индуцируется разворачиванием одного из белков в димере при активации. В димерах, состоящих из двух почти идентичных белков, белок с более высокой конформационной гибкостью отделяется от комплекса с большим зарядом. Это может быть связано с тем, что часть молекулы покидает барьер диссоциации, где дальнейшее развертывание молекулы индуцируется отталкивающим кулоновским потенциалом.Во время этого процесса разворачивания и разделения протоны передаются разворачивающемуся белку, чтобы уменьшить общую кулоновскую энергию. Этот механизм лучше всего объясняет результаты, наблюдаемые для гетеродимеров α -лактабумина, состоящих из одного окисленного и одного восстановленного белка. Этот механизм также лучше всего объясняет результаты для димеров цитохрома c с 11 зарядами, для которых наблюдается энергетическая зависимость в степени асимметрии [56].
Некоторая часть наблюдаемого асимметричного распределения заряда также может быть приписана другому механизму, в котором структуры мономеров в димере различаются.Это лучше всего объясняет результаты, наблюдаемые для димеров цитохрома c с зарядами 17+ по сравнению с димерами с 13+ зарядами. При образовании непосредственно из раствора первый диссоциирует с симметричным распределением заряда, тогда как последний диссоциирует с сильно асимметричным распределением заряда. Однако, когда зарядовое состояние ионов димера 17+ снижается до 13+ в газовой фазе посредством мягких реакций переноса протона, образующиеся ионы 13+ диссоциируют с симметричным распределением заряда.Это ясно демонстрирует, что ионы 13+, образованные из ионов 17+, имеют другую структуру (или разные ансамбли структур), чем те, которые образуются непосредственно из раствора, и что это структурное различие отвечает за асимметричное и симметричное распределение заряда, наблюдаемое при диссоциации этих 13+. ионы. Это первое зарегистрированное свидетельство того, что асимметричное распределение заряда может быть вызвано структурными различиями в субъединицах мономера в комплексе.
Наконец, H / D обмен зарядового состояния 11+ димеров цитохрома c указывает на присутствие множественных конформеров, хотя эти конформеры не разрешаются в этом эксперименте.Дальнейшая работа с другими реагентами для обмена H / D или в других условиях, или с конформационно-селективными методами, такими как подвижность ионов, в сочетании с экспериментами по диссоциации может предоставить дополнительную полезную информацию о процессе разделения заряда. Лучшее понимание этого процесса в конечном итоге улучшит структурную информацию, которая может быть получена при газофазной диссоциации нековалентных комплексов.
Благодарности
Авторы благодарят доктора Дэвида Кинга (Калифорнийский университет, Беркли) за восстановление и очистку α -лактальбумина.Эта работа была поддержана Национальным институтом здравоохранения (грант № R01-GM64712-01).
Ссылки
1. Калета Д.Т., Джарролд М.Ф. Пептидные вертушки. J Am Chem Soc. 2002; 124: 1154–1155. [PubMed] [Google Scholar] 2. Kaleta DT, Jarrold MF. Нековалентные взаимодействия между несольватированными пептидами. J. Phys Chem A. 2002; 106: 9655–9664. [Google Scholar] 3. Clemmer DE, Jarrold MF. Измерения подвижности ионов и их приложения к кластерам и биомолекулам. J. Mass Spectrom. 1997. 32: 577–592. [Google Scholar] 4.Counterman AE, Valentine SJ, Srebalus CA, Henderson SC, Hoaglund CS, Clemmer DE. Структура высокого порядка и диссоциация газообразных пептидных агрегатов, скрытых в масс-спектрах. J Am Soc масс-спектрометрия. 1998. 9: 743–759. [PubMed] [Google Scholar] 5. Counterman AE, Hilderbrand AE, Barnes CAS, Clemmer DE. Формирование пептидных агрегатов во время ESI: размер, заряд, состав и вклад в шум. J Am Soc масс-спектрометрия. 2001; 12: 1020–1035. [Google Scholar] 6. Гидден Дж., Виттенбах Т., Батька Дж. Дж., Вайс П., Джексон А. Т., Скривенс Дж. Х., Бауэрс МТ.Энергетика сворачивания и динамика макромолекул в газовой фазе: щелочные ионно-катионизированные олигомеры поли (этилентерефталата). J Am Chem Soc. 1999; 121: 1421–1422. [Google Scholar] 7. Гевремонт Р., Purves RW. Спектрометрия-масс-спектрометрия подвижности ионов с асимметричной формой волны в высоком поле: исследование ионов лейцина-энкефалина, полученных с помощью ионизации электрораспылением. J Am Soc масс-спектрометрия. 1999; 10: 492–501. [PubMed] [Google Scholar] 8. Purves RW, Barnett DA, Guevremont R. Разделение белков-конформеров с использованием спектрометрии-масс-спектрометрии асимметричной формы волны с асимметричной формой волны электроспрея.Int J масс-спектрометрия. 2000; 197: 163–177. [Google Scholar] 9. Дэн Ю.З., Чжан З.К., Смит Д.Л. Сравнение непрерывного и импульсного мечения амидного водородного обмена / масс-спектрометрии для исследования динамики белков. J Am Soc масс-спектрометрия. 1999; 10: 675–684. [PubMed] [Google Scholar] 10. Чжу М.М., Ремпель Д.Л., Брутто М.Л. Данные моделирования титрования, амидного H / D обмена и масс-спектрометрии для получения констант связывания белок-лиганд. J Am Soc масс-спектрометрия. 2004. 15: 388–397. [PubMed] [Google Scholar] 11. Гарда Э., Уилларда Д., Брегара Дж., Грин МК, Лебрилла CB.Сайт-специфичность в H-D-обменных реакциях газофазных протонированных аминокислот с Ch4OD. Org Mass Spectrom. 1993; 28: 1632–1639. [Google Scholar] 12. Кэмпбелл С., Роджерс М. Т., Марцлафф Е. М., Бошам Дж. Л.. Структурные и энергетические ограничения на реакции газофазного водородно-дейтериевого обмена протонированных пептидов с D2O, CD3OD, CD3CO2D и ND3. J Am Chem Soc. 1994; 116: 9765–9766. [Google Scholar] 13. Gard E, Green MK, Bregar J, Lebrilla CB. Газофазный водород-дейтериевый обмен как молекулярный зонд для взаимодействия метанола и протонированных пептидов.J Am Soc масс-спектрометрия. 1994; 5: 623–631. [PubMed] [Google Scholar] 14. Кэмпбелл С., Роджерс М. Т., Марцлафф Е. М., Бошам Дж. Л.. Реакции обмена дейтерия как зонд структуры биомолекул. Фундаментальные исследования газофазных реакций H / D обмена протонированных олигомеров глицина с D2O, CD3OD, CD3CO2D и ND3. J Am Chem Soc. 1995; 117: 12840–12854. [Google Scholar] 15. Грин МК, Penn SG, Lebrilla CB. Комплексообразование протонированных пептидов с сахаридами в газовой фазе снижает скорость реакций обмена водород / дейтерий.J Am Soc масс-спектрометрия. 1995; 6: 1247–1251. [PubMed] [Google Scholar] 16. Калташов И.А., Дорошенко В.М., Коттер Р.Дж. Газофазные реакции обмена пептидных ионов водород / дейтерий в квадрупольном масс-спектрометре с ионной ловушкой. Белки. 1997. 28: 53–58. [PubMed] [Google Scholar] 17. Heck AJR, Jorgensen TJD, O’Sullivan M, von Raumer M, Derrick PJ. Газофазные нековалентные взаимодействия между антибиотиками группы ванкомицина и пептидами-предшественниками бактериальной клеточной стенки, вызванные обменом водорода / дейтерия. J Am Soc масс-спектрометрия.1998. 9: 1255–1266. [Google Scholar] 18. Фрейтас М.А., Маршалл АГ. Скорость и степень газофазного водородно-дейтериевого обмена брадикининов: данные о пептидных цвиттерионах в газовой фазе. Int J масс-спектрометрия. 1999. 183: 221–231. [Google Scholar] 19. Рейзер ML, Brodbelt JS. Газофазные реакции обмена H / D полиаминовых комплексов: [M + H) (+), (M плюс щелочной металл (+)] и (M + 2H) (2+) J Am Soc Mass Spectrom. 2000; 11 : 711–721. [PubMed] [Google Scholar] 20. Леви-Сери Э, Костер Г., Коган А., Гутман К., Рубен Б. Г., Лифшиц К.Исследование H / D-обмена в протонированном брадикинине с помощью ионизационной трубки с электрораспылением. J. Phys Chem A. 2001; 105: 5552–5559. [Google Scholar] 21. Солоуки Т., Форт Р.С., Аломари А., Фаттахи А. Реакции газофазного водородно-дейтериевого обмена модельного пептида: FT-ICR и вычислительный анализ конформационных мутаций, индуцированных металлами. J Am Soc масс-спектрометрия. 2001; 12: 1272–1285. [PubMed] [Google Scholar] 22. Suckau D, Shi Y, Beu SC, Senko MW, Quinn JP, Wampler FM, McLafferty FW. Сосуществующие стабильные конформации газообразных белковых ионов.Proc Nat Acad Sci USA. 1993; 90: 790–793. [Бесплатная статья PMC] [PubMed] [Google Scholar] 23. Wood TD, Chorush RA, Wampler FM, Little DP, O’Connor PB, McLafferty FW. Газофазное сворачивание и разворачивание катионов цитохрома- c . Proc Natl Acad Sci USA. 1995; 92: 2451–2454. [Бесплатная статья PMC] [PubMed] [Google Scholar] 24. McLafferty FW, Guan ZQ, Haupts U, Wood TD, Kelleher NL. Газообразные конформационные структуры цитохрома c. J Am Chem Soc. 1998. 120: 4732–4740. [Google Scholar] 25. Рид Г.Е., О’Хэр RAJ, Styles ML, McFadyen WD, Simpson RJ.Газофазные ионно-молекулярные реакции в модифицированной ионной ловушке: H / D-обмен нековалентных комплексов и координационно-ненасыщенных комплексов платины. Масс-спектрометр Rapid Commun. 1998; 12: 1701–1708. [Google Scholar] 26. Коган А., Устюжанин П., Рубен Б. Г., Лифшиц С. Водород / дейтериевый обмен мономеров и димеров лейцин-энкефалина. Int J масс-спектрометрия. 2002; 213: 1–4. [Google Scholar] 27. Мао Д., Дуглас ди-джей. H / D-обмен газофазными ионами брадикинина в линейной квадрупольной ионной ловушке. J Am Soc масс-спектрометрия.2003. 14: 85–94. [PubMed] [Google Scholar] 28. Ли SW, Ли Х.Н., Ким Х.С., Бошам Дж. Л.. Селективное связывание краун-эфиров с протонированными пептидами может быть использовано для исследования механизмов обмена H / D и реакций диссоциации, вызванных столкновениями, в газовой фазе. J Am Chem Soc. 1998. 120: 5800–5805. [Google Scholar] 29. Валовой DS, Schnier PD, Rodriguezcruz SE, Fagerquist CK, Williams ER. Конформации и сворачивание ионов лизоцима в вакууме. Proc Nat Acad Sci USA. 1996; 93: 3143–3148. [Бесплатная статья PMC] [PubMed] [Google Scholar] 31.Loo JA. Изучение нековалентных белковых комплексов методом масс-спектрометрии с ионизацией электрораспылением. Масс-спектром. Ред. 1997; 16: 1–23. [PubMed] [Google Scholar] 32. Лим Х.К., Сие Й.Л., Ганем Б., Хенион Дж. Распознавание пептидных лигандов клеточной стенки антибиотиками группы ванкомицина – исследования с использованием масс-спектрометрии с ионным распылением. J. Mass Spectrom. 1995; 30: 708–714. [Google Scholar] 33. Лоо Дж. А., Ху П. Ф., МакКоннелл П., Мюллер В. Т., Сойер Т. К., Танабал В. Исследование взаимодействий связывания белка с фосфопептидом домена Src Sh3 с помощью масс-спектрометрии с ионизацией электрораспылением.J Am Soc масс-спектрометрия. 1997. 8: 234–243. [Google Scholar] 34. Йоргенсен TJD, Roepstorff P, Heck AJR. Прямое определение констант связывания раствора для нековалентных комплексов между пептидными аналогами бактериальной клеточной стенки и антибиотиками группы ванкомицина методом масс-спектрометрии с ионизацией электрораспылением. Anal Chem. 1998; 70: 4427–4432. [Google Scholar] 35. Дэниел Дж. М., Фрисс С. Д., Раджагопалан С., Вендт С., Зеноби Р. Количественное определение нековалентных связывающих взаимодействий с использованием масс-спектрометрии с мягкой ионизацией.Int J масс-спектрометрия. 2002; 216: 1-27. [Google Scholar] 36. Валовой DS, Чжао YX, Уильямс ER. Диссоциация комплексов гем-глобин с помощью инфракрасной радиационной диссоциации черного тела: молекулярная специфичность в газовой фазе? J Am Soc масс-спектрометрия. 1997. 8: 519–524. [Бесплатная статья PMC] [PubMed] [Google Scholar] 37. Schnier PD, Klassen JS, Strittmatter EE, Williams ER. Энергии активации для диссоциации двухцепочечных олигонуклеотидных анионов: данные о спаривании оснований Уотсона-Крика в вакууме. J Am Chem Soc. 1998; 120: 9605–9613.[Бесплатная статья PMC] [PubMed] [Google Scholar] 38. Рогалевич Ф., Хоппиллиард Я., Оханесян Г. Структуры и фрагментация комплексов цинка (II) аминокислот в газовой фазе. IV. Влияние растворителя на структуру электрораспыленных ионов. Int J масс-спектрометрия. 2003. 227: 439–451. [Google Scholar] 39. Габелика В., Де Пау Э. Сравнение между фазовой стабильностью в растворе и кинетической стабильностью в газовой фазе олигодезоксинуклеотидных дуплексов. J. Mass Spectrom. 2001; 36: 397–402. [PubMed] [Google Scholar] 40. Габелика V, Де Пау Э.Сравнение индуцированной столкновением диссоциации дуплексной ДНК при различных режимах коллизии: доказательства многоступенчатого механизма диссоциации. J Am Soc масс-спектрометрия. 2002; 13: 91–98. [PubMed] [Google Scholar] 41. Габелика В., Де Пау Э. Диссоциация 16-мерных дуплексов ДНК с различными последовательностями, вызванная столкновением: свидетельства сохранения конформации двойной спирали в газовой фазе. Int J масс-спектрометрия. 2002; 219: 151–159. [Google Scholar] 42. Ван К.Х., Шибуэ Т., Брутто ML. Нековалентные комплексы между ДНК-связывающими лекарствами и двухцепочечными олигодезоксинуклеотидами: исследование методом масс-спектрометрии с ионной ловушкой ESI.J Am Chem Soc. 2000; 122: 300–307. [Google Scholar] 43. Чен Ю.Л., Кэмпбелл Дж. М., Коллингс Б. А., Конерманн Л., Дуглас Д. Д.. Устойчивость высокозаряженного нековалентного комплекса в газовой фазе: голомиоглобина. Масс-спектрометр Rapid Commun. 1998. 12: 1003–1010. [PubMed] [Google Scholar] 44. Хантер С.Л., Маук А.Г., Дуглас Диджей. Диссоциация гема от миоглобина и цитохрома b (5): сравнение поведения в растворе и газовой фазе. Биохим США. 1997; 36: 1018–1025. [PubMed] [Google Scholar] 45. Йоргенсен Т.Д., Делфорж Д., Ремакл Дж., Бойсен Дж., Рёпсторф П.Индуцированная столкновением диссоциация нековалентных комплексов между антибиотиками ванкомицина и стереоизомерами пептидного лиганда: доказательства молекулярного распознавания в газовой фазе. Int J масс-спектрометрия. 1999. 188: 63–85. [Google Scholar] 46. Loo JA, He JX, Cody WL. Структура высшего порядка в газовой фазе отражает структуру раствора. J Am Chem Soc. 1998; 120: 4542–4543. [Google Scholar] 47. Ростом А.А., Фучини П., Бенджамин Д.Р., Юенеманн Р., Нирхаус К.Х., Хартл Ф.У., Добсон С.М., Робинсон К.В. Обнаружение и селективная диссоциация интактных рибосом в масс-спектрометре.Proc Nat Acad Sci USA. 2000; 97: 5185–5190. [Бесплатная статья PMC] [PubMed] [Google Scholar] 48. Pinkse MWH, Maier CS, Kim JI, Oh BH, Heck AJR. Макромолекулярная оценка уреазы Helicobacter pylori, исследованная методом масс-спектрометрии. J. Mass Spectrom. 2003. 38: 315–320. [PubMed] [Google Scholar] 49. Лайт-Вал К.Дж., Шварц Б.Л., Смит Р.Д. Наблюдение нековалентных четвертичных ассоциаций белков с помощью масс-спектрометрии с ионизацией электрораспылением. J Am Chem Soc. 1994; 116: 5271–5278. [Google Scholar] 50. Шварц Б.Л., Брюс Дж. Э., Андерсон Г. А., Хофстадлер С. А., Роквуд А. Л., Смит Р. Д., Чилкоти А., Стейтон П. С..Диссоциация тетрамерных ионов нековалентных стрептавидиновых комплексов, образующихся при ионизации электрораспылением. J Am Soc масс-спектрометрия. 1995; 6: 459–465. [PubMed] [Google Scholar] 51. Фицджеральд М.С., Чернушевич И., Стэндинг К.Г., Уитмен С.П., Кент SBH. Исследование олигомерной структуры фермента методом времяпролетной масс-спектрометрии с ионизацией электрораспылением. Proc Nat Acad Sci USA. 1996; 93: 6851–6856. [Бесплатная статья PMC] [PubMed] [Google Scholar] 52. Фелицын Н., Китова Е.Н., Классен Я.С. Термическое разложение газообразного мультибелкового комплекса, исследованное методом черной инфракрасной радиационной диссоциации.Исследование происхождения асимметричного поведения диссоциации. Anal Chem. 2001; 73: 4647–4661. [PubMed] [Google Scholar] 53. Бенеш JLP, Соботт Ф., Робинсон CV. Термическая диссоциация мультимерных белковых комплексов с помощью масс-спектрометрии с наноэлектрораспылением. Anal Chem. 2003. 75: 2208–2214. [PubMed] [Google Scholar] 55. Versluis C, van der Staaij A, Stokvis E, Heck AJR, de Craene B. Образование метастабильных ионов и разделение разрозненных зарядов при газофазной диссекции белковых сборок, изученных методом ортогональной времяпролетной масс-спектрометрии.J Am Soc масс-спектрометрия. 2001; 12: 329–336. [PubMed] [Google Scholar] 56. Jurchen JC, Williams ER. Происхождение асимметричного разделения заряда при диссоциации газофазных белковых гомодимеров. J Am Chem Soc. 2003; 125: 2817–2826. [Бесплатная статья PMC] [PubMed] [Google Scholar] 57. Jurchen JC, Garcia DE, Williams ER. Пути газофазной диссоциации многозарядных пептидных кластеров. J Am Soc масс-спектрометрия. 2003. 14: 1373–1386. [Бесплатная статья PMC] [PubMed] [Google Scholar] 58. Готье Дж. В., Траутман Т. Р., Якобсон Д.Б.Продолжительное нерезонансное облучение для диссоциации, активируемой столкновениями, с использованием метода масс-спектрометрии с преобразованием Фурье и диссоциации, активируемой столкновениями, которая имитирует инфракрасную многофотонную диссоциацию. Анальный Чим Акта. 1991; 246: 211–225. [Google Scholar] 59. Вингер Б.Э., Лайт-Валь КДЖ, Роквуд А.Л., Смит Р.Д. Исследование качественных различий конформации многократно протонированных газофазных белков с помощью изотопного обмена H / D с D2O. J Am Chem Soc. 1992; 114: 5897–5898. [Google Scholar] 60. Фелицын Н., Китова Е.Н., Классен Я.С.Термическая диссоциация гомодимера белка экотина в газовой фазе. J Am Soc масс-спектрометрия. 2002; 13: 1432–1442. [PubMed] [Google Scholar] 61. Апостол I. Оценка относительной стабильности сконструированных гемоглобинов с помощью масс-спектрометрии с электрораспылением. Анальная биохимия. 1999; 272: 8–18. [PubMed] [Google Scholar] 62. Versluis C, Heck AJR. Газофазная диссоциация гемоглобина. Int J масс-спектрометрия. 2001; 210: 637–649. [Google Scholar] 63. Цена WD, Williams ER. Активация пептидных ионов излучением черного тела: факторы, приводящие к кинетике диссоциации в пределе быстрого обмена энергией.J. Phys Chem A. 1997; 101: 8844–8852. [Бесплатная статья PMC] [PubMed] [Google Scholar]Дальнейшие исследования происхождения асимметричного разделения заряда в белковых гомодимерах
J Am Soc Mass Spectrom. Авторская рукопись; доступно в PMC 2006 6 января.
Опубликован в окончательной редакции как:
PMCID: PMC1325214
NIHMSID: NIHMS3986
Химический факультет Калифорнийского университета в Беркли, Беркли, Калифорния, США
Запросы на перепечатку адресов: ДокторЭ. Р. Уильямс, химический факультет Калифорнийского университета в Беркли, Беркли, Калифорния 94720, США. Электронное письмо: ude.yelekreb.mehcc@smailliw См. другие статьи в PMC, в которых цитируется опубликованная статья.Abstract
Диссоциация протонированных в газовой фазе белковых димеров на составляющие их мономеры может приводить к симметричному или асимметричному распределению заряда. Диссоциация гомодимеров α -лактальбумина с 15+ зарядами приводит к симметричному, но широкому распределению белковых мономеров с зарядовыми состояниями, сосредоточенными вокруг 8 + / 7 +.Напротив, диссоциация гетеродимера 15+, состоящего из одной молекулы в окисленной форме и одной в восстановленной форме, приводит к сильно асимметричному распределению зарядов, при котором восстановленные частицы уносят преимущественно заряды 11+, а окисленная молекула уносит заряды 4+. . Этот результат не может быть адекватно объяснен дифференциальной зарядкой, происходящей ни в растворе, ни в процессе электрораспыления, но, по-видимому, лучше всего объясняется восстановленными частицами, разворачивающимися при активации в газовой фазе с последующим разделением и переносом протонов на разворачивающиеся частицы в диссоциативном комплексе. чтобы минимизировать кулоновское отталкивание.Для димеров цитохрома c , образованных непосредственно из раствора, зарядовое состояние 17+ подвергается симметричному распределению заряда, тогда как диссоциация 13+ асимметрична. Уменьшение зарядового состояния димеров с зарядами 17+ до 13+ посредством переноса протона в газовой фазе и последующей диссоциации выбранных по массе ионов 13+ приводит к симметричному распределению заряда. Этот результат ясно показывает, что структура димерных ионов с зарядами 13+ зависит от метода образования иона и что структурные различия ответственны за наблюдаемое симметричное и асимметричное разделение зарядов.Это указывает на то, что асимметрия, наблюдаемая, когда эти ионы образуются непосредственно из раствора, должна происходить либо из-за различий в конформациях мономера в димере, которые существуют в растворе, либо в процессе ионизации электрораспылением. Эти результаты предоставляют дополнительные доказательства происхождения асимметрии заряда, которая возникает при диссоциации многозарядных белковых комплексов, и показывают, что некоторая информация о фазе раствора может быть получена из этих экспериментов по газофазной диссоциации.
Биологическая функциональность белков и белковых комплексов сильно зависит от их структур в фазе раствора. Важность области структурной биологии отражена в нескольких недавно присужденных Нобелевских премиях за вклад в методы структурного выяснения, включая ЯМР (Курт Вутрих, 2002 г. по химии), рентгеновскую кристаллографию (Иоганн Дайзенхофер, Роберт Хубер и Хартмут Мишель, 1988 г. в. химия; Герберт Хауптман и Джером Карл 1985 по химии) и кристаллографическая электронная микроскопия (сэр Аарон Клаг 1982 по химии).Анализ белков и белковых комплексов этими методами может предоставить подробную трехмерную информацию, но эти методы часто ограничены экспериментальными ограничениями, такими как необходимость получения полезных кристаллов белка или требование значительного количества образца. Напротив, для масс-спектрометрии требуется небольшой образец, а с ионизацией электрораспылением (ESI) или мягкой лазерной десорбцией / ионизацией (John Fenn and Kiochi Tanaka, 2002 по химии) в масс-спектрометр можно ввести большое количество биомолекул и специфических комплексов.Был разработан ряд методов для исследования общих трехмерных форм ионов биомолекул и нековалентных комплексов с использованием методов ионной подвижности [1–8], H / D обмена как в растворе [9, 10], так и в газовой фазе [ 11–28] и реакционной способности с переносом протона [29, 30].
Для анализа нековалентных комплексов масс-спектрометрия имеет то преимущество, что стехиометрию комплекса можно определить с помощью простого измерения массы [31]. В некоторых случаях MS может обеспечить правильные константы связывания в фазе раствора для белок-лигандных взаимодействий [32–35].Эти разработки подробно рассмотрены [31, 35]. В принципе, информацию о структуре нековалентных комплексов можно потенциально получить, исследуя ионы продуктов, образующиеся в результате диссоциации комплекса, в эксперименте тандемной масс-спектрометрии (МС / МС). Объем соответствующей информации, которую можно получить в результате этих экспериментов, зависит от количества структурной информации о фазе раствора, которая сохраняется биологическими комплексами при переходе в газовую фазу (или от того, может ли эта информация быть восстановлена на основе сведений о происходящих структурных изменениях. во время этого перехода) и степень, в которой методы MS / MS нарушают любую оставшуюся структурную информацию.Многие эксперименты показывают, что газофазные комплексы биомолекул и даже некоторые небольшие молекулы могут сохранять «память» о своей фазовой структуре раствора, и что эта память может быть исследована процессами диссоциации [36–48].
Ряд исследований гомогенных биологических комплексов (тетрамеры, пентамеры, гексамеры и т. Д.) С помощью МС / МС выявили первоначально неожиданный результат, заключающийся в том, что первичный путь диссоциации мультимерных комплексов представляет собой выброс высоко заряженного мономера, который удаляет 30-50 % заряда комплекса [49–53].Рентгеновская кристаллография показывает, что структуры растворов субъединиц во многих гомогенных комплексах идентичны. В некоторых случаях весь комплекс кристаллизуется внутри асимметричной единицы кристалла, как в случае с шига-подобным токсином [9], в то время как в других, таких как апострептавидин, асимметричная единица содержит одну субъединицу, и структура комплекс экстраполирован с помощью молекулярного моделирования [54]. Из-за практически идентичных структур фазы раствора участвующих субъединиц в этих биологических комплексах наблюдаемое асимметричное распределение заряда, по-видимому, является артефактом процедуры ESI-MS / MS.Было высказано предположение, что асимметричное распределение заряда коррелирует с газофазными конформационными перестройками [50, 52, 55, 56]. Также было продемонстрировано, что степень асимметричного распределения заряда для гомодимеров белков зависит от конформационной гибкости белков в комплексе, зарядового состояния комплекса, отложения внутренней энергии и состава растворов для электрораспыления, из которых комплексы образуются. формируются [56]. Для более крупных кластеров, состоящих из идентичных субъединиц, наблюдалось деление с преимущественным образованием протонированных димеров, а с увеличением зарядового состояния и размера кластера наблюдалось усиление образования протонированного мономера [57].Степень деления по сравнению с потерями нейтралов для этих кластеров может быть связана с наблюдаемой для многозарядных металлических кластеров с одним важным отличием: различия в конформационной гибкости субъединиц мономера внутри кластера приводят к различиям в коэффициентах ветвления различных диссоциаций. процессы, наблюдаемые для этих многократно протонированных кластеров биомолекул [57].
Газофазный водород-дейтериевый (H / D) обмен обычно осуществляется путем хранения интересующего иона в вакууме и измерения изменения массы иона из-за реакций обмена H / D, которые происходят во время столкновений с нейтральными молекулами обменного реагента. , например, D 2 O или ND 3 .Хотя газофазный обмен H / D широко использовался для характеристики пептидов и малых молекул [11–21], а также структуры некоторых газофазных белков [22–24], очень мало исследований было выполнено с нековалентными комплексами, включающими биологические ионы, и эта нековалентная работа была сосредоточена на димерах пептидов [25, 26] и пептидных комплексах с ионами металлов [16, 18, 21, 27] и небольшими молекулами [15, 28].
Здесь представлена дальнейшая характеристика гомодимеров белка с использованием газофазной диссоциации и газофазного H / D обмена.Исследования диссоциации смешанных димеров окисленного и восстановленного α -лактальбумина предоставляют дополнительные доказательства того, что конформационная гибкость играет ключевую роль в определении степени асимметричного распределения заряда. Для диссоциации гомодимеров цитохрома c показано, что зарядовые состояния, полученные в результате снижения заряда в газовой фазе из гомодимеров с более высоким зарядовым состоянием, диссоциируют совсем иначе, чем состояния, полученные непосредственно из раствора. Представлена первая демонстрация газофазного H / D обмена белкового комплекса.Эти результаты показывают, что два разных механизма ответственны за асимметричное распределение заряда, наблюдаемое при диссоциации гомодимеров белка.
Experimental
Эксперименты проводились на масс-спектрометре Berkeley-Bruker с узким проходом 9,4 Тл с ионно-циклотронным резонансом с преобразованием Фурье (FT-ICR), который описан в другом месте [56]. Используемые экспериментальные процедуры аналогичны использованным ранее [56]. Краткое описание и дополнительные сведения приведены ниже.
Chemicals
Цитохром сердца лошади c , α -лактальбумин из коровьего молока и триэтиламин были приобретены у Sigma-Aldrich Co. (Сент-Луис, Миссури) и использовались без дополнительной очистки. Дейтерированный аммиак был приобретен в Cambridge Isotope Laboratories (Woburn, MA). Ацетамидирование α -лактальбумина было выполнено доктором Дэвидом Кингом (Калифорнийский университет, Беркли). Все четыре внутримолекулярные дисульфидные связи α -лактальбумина восстанавливаются трис (карбоксиэтил) фосфином, ацетамидируются в темноте йодацетамидом в течение одного часа и очищаются с помощью ВЭЖХ для удаления буфера и непрореагировавших реагентов.Практически полное восстановление и ацетамидирование всех дисульфидных связей в α -лактальбумине было подтверждено измерениями массы.
Образование димеров
Образование неспецифических белковых димеров усиливается за счет использования концентраций белка 100–500 мкМ M и регулирования напряжений на границе раздела для «мягкого» введения ионов. Сигнал димера улучшается за счет настройки на селективное накопление ионов во внешнем гексаполе в области источника прибора. Это достигается накоплением ионов в гексаполе в течение 3–4 с (против ≤ 1.0 с), увеличивая смещение постоянного тока гексаполя с ~ 2,7 В до 3,5–4,5 В и вводя несколько скоплений гексаполя в ионную ячейку перед обнаружением.
Смешанные димеры окисленного (все четыре дисульфидные связи не повреждены) и восстановленного (все четыре дисульфидные связи разорваны и все восемь остатков цистеина впоследствии ацетамидированы) α -лактальбумин были образованы путем смешивания растворов двух форм α -лактальбумина в различных соотношения. Концентрация общего белка поддерживалась постоянной на уровне ~ 200 мкМ М, и отношения двух компонентов были изменены, чтобы максимизировать абсолютное содержание смешанного димера.Наибольшее количество смешанного димера появляется при относительной концентрации восстановленного до окисленного α -лактальбумина ~ 2: 1. Поскольку концентрации белка определялись массой лиофилизированных порошков, используемых для приготовления раствора, эти концентрации являются приблизительными. Ионы димера белка формировали с использованием ESI со скоростью потока ~ 1 мкМ л / мин в условиях раствора 1: 1 вода: метанол + 2% уксусная кислота.
Димеры цитохрома c были сформированы с использованием обычного ESI при скорости потока 1-2 мкМ л / мин и концентрации 100 мкМ М в 1: 1 вода: метанол + 2% уксусная кислота.Зарядовые состояния димеров, сформированные непосредственно из раствора, варьируются от [(цитохром c ) 2 + 19H] 19+ до [(циктохром c ) 2 + 13H] 13+ (сокращенно D19 – D13) . Зарядовые состояния димера с m / z выше, чем у D17, были выброшены из ионной ячейки с использованием коррелированных выстрелов, оставляя зарядовые состояния D19 – D17, оставшиеся в ионной ячейке. Вакуумная камера FT-ICR была подготовлена для снижения заряда путем кратковременного введения триэтиламина в вакуумную камеру под давлением ~ 3 × 10 90 · 10 7 -8 90 · 108 торр с использованием пьезоэлектрического импульсного клапана.Импульсный клапан затем закрывался через 5 с, и базовое давление прибора возвращалось к ~ 8 × 10 90 · 107 -9 90 · 108 торр через 95 с. Снижение заряда посредством переноса протона осуществлялось путем улавливания гомодимеров цитохрома c с зарядовыми состояниями D19 – D17 в приборе до тех пор, пока столкновения с остаточными молекулами триэтиламина не снижали зарядовое состояние димеров в течение 100 с, так что 13+ было самая распространенная форма димера. Вновь образованные ионы D13 были изолированы с использованием коррелированных свип-сигналов и диссоциированы с помощью длительной диссоциации, активируемой столкновениями (SORI-CAD) [58] вне резонанса и вне резонанса, при +600 Гц вне резонанса и 3.5 В пик – пик в течение 0,25 с после подачи импульса азота, повышающего давление в вакуумной камере до ~ 10 –6 торр.
Газофазный H / D обмен белковых комплексов
Газофазный H / D обмен гомодимеров цитохрома c осуществляли путем улавливания всех генерируемых электрораспылением ионов в ячейке и введения ND 3 в вакуумную камеру с использованием ручной герметичный клапан (Varian, Lexington, MA) на 180 с при нескольких давлениях в диапазоне от ~ 1 × 10 −8 до максимального ~ 3 × 10 −7 торр.После 180 с замены клапан утечки был закрыт, и вакуумная камера откачивалась в течение 60–120 с, пока базовое давление не стало достаточно низким для изотопного разрешения (<1 × 10 –8 торр). Предыдущие эксперименты по газофазному обмену H / D с белками использовали D 2 O в качестве обменного реагента [22–24, 59]. Из-за трудностей, связанных с длительным временем улавливания (> 300 с) и значительным временем, необходимым для удаления относительно нелетучего D 2 O, в качестве обменного реагента был выбран ND 3 .Давление вводимого в вакуумную камеру НД 3 измерялось некалиброванным ионным манометром, расположенным на расстоянии ~ 1,1 м от ионной ячейки.
Результаты и обсуждение
Диссоциация смешанных
димеров α -лактальбуминаИспользуя растворы ESI с высокими концентрациями белка, можно образовать как гомодимеры окисленного белка α -лактальбумина (каждый белок имеет четыре интактных дисульфидных связи), так и окисленно-восстановленные гетеродимеры α -лактальбумина (один белок имеет четыре интактных дисульфидных связи, а другой – четыре восстановленные дисульфидные связи, причем все восемь остатков цистеина ацетамидированы).И окисленные гомодимеры, и окисленно-восстановленные гетеродимеры образуются непосредственно из ESI с 15 присоединенными протонами ((Ox-Ox) 15 и (Ox-Red) 15, соответственно) (). (Ox-Ox) 15 диссоциирует с довольно широким, но симметричным распределением заряда (), аналогичным тому, что наблюдалось ранее для (Ox-Ox) 13 и (Red-Red) 17 [56]. (Red-Red) 13, как было ранее показано, асимметрично диссоциирует с преимущественным образованием M9 / M4 [56]. В отличие от (Ox-Ox) 15, (Ox-Red) 15 диссоциирует с сильно асимметричным распределением заряда ().Наиболее распространенным процессом диссоциации димера является (Ox-Red) 15 → Red11 + Ox4, процесс разделения, при котором восстановленный α -лактальбумин удаляет почти всего заряда! Обратите внимание, что большая интенсивность сигнала Red11 по сравнению с Ox4 обусловлена как более низким уровнем заряда последнего (сигнал FT-ICR пропорционален заряду), так и несколько меньшей эффективностью обнаружения при более высоком m / z .
( a ) Масс-спектр ESI, полученный из 200 мкМ раствора M (общая концентрация белка), содержащего окисленный (нетронутый дисульфид) и восстановленный α -лактальбумин (1: 1 вода: метанол + 2% уксусная кислота) , ( b ) МС / МС спектр окисленного-восстановленного гетеродимера α -лактальбумина с 15 протонами, (Ox-Red) 15 и ( c ) МС / МС спектр окисленного α -лактальбумина -окисленный гомодимер (Ox-Ox) 15, образованный из раствора ~ 125 мкМ М окисленного α -лактальбумина в 1: 1 вода: метанол + 2% уксусная кислота.Red8 представляет (восстановленный α -лактальбумин + 8H) 8+ и т. Д.
Происхождение сильно асимметричного распределения заряда может быть связано с асимметричным разделением, которое происходит в газовой фазе [56], или с асимметрией конформации или заряд мономеров, составляющих димер, либо в растворе, либо индуцированный процессами электрораспыления. В первом механизме асимметричное распределение заряда происходит из-за конформационного изменения одной из субъединиц мономера, которая составляет димер, после активации.Когда мономер разворачивается, молекула преодолевает активационный барьер для разворачивания и разделения. Из-за кулоновской энергии выход из этого барьера является сильно отталкивающим, и когда одна часть разворачивающейся молекулы выходит из этого канала, она будет управлять разворачиванием остальной части молекулы, когда она отделяется от другой субъединицы мономера. В этом процессе протоны будут переходить к выходящему разворачивающемуся иону, чтобы минимизировать кулоновскую энергию [30]. Поскольку восстановленный белок более гибок конформационно и может разворачиваться в значительно большей степени, чем окисленный белок, восстановленный белок разворачивается и уносит больший заряд, чем окисленная форма.
Альтернативный механизм этой асимметрии заряда заключается в том, что (Ox-Red) 15 может образовываться в растворе путем агрегации сильно заряженных восстановленных и низкозарядных окисленных мономеров α -лактальбумина в растворе или образовываться во время электрораспыления за счет асимметричной зарядки. отдельных белковых составляющих на заключительных этапах ESI [60]. Также было высказано предположение, что асимметричное распределение заряда для гетеродимеров может быть связано с различиями в pI [61, 62]. Хотя возникает разница в зарядке этих двух молекул, она не учитывает величину асимметричного разделения заряда.В масс-спектре ESI смешанного раствора α -лактальбумина восстановленный α -лактальбумин появляется преимущественно при 8+ и в меньшей степени при 9+ и 10+ (). Наблюдается очень мало 11+. Окисленный α -лактальбумин встречается только в зарядовом состоянии 8+. В спектре диссоциации (Ox-Red) 15 доминирующими наблюдаемыми видами являются Red11 и Ox5, несмотря на то, что численность этих видов, продуцируемых непосредственно ESI, незначительна. Хотя можно было бы рационализировать более низкое зарядовое состояние, наблюдаемое для Ox, как немного более низкое зарядовое состояние димера по сравнению с суммой зарядовых состояний отдельных частиц, это не может быть верным для восстановленных частиц, которые имеют значительно более высокий заряд при диссоциации. спектра, чем при формировании непосредственно электрораспылением.Таким образом, этот результат трудно рационализировать, основываясь только на модели, в которой асимметричное распределение заряда является либо реликтом агрегации фазы раствора, либо артефактом процесса электрораспыления. Результаты гетеродимеров (Ox-Red) 15 α -лактальбумина, по-видимому, лучше всего подтверждают процесс асимметричного распределения заряда в газовой фазе [56].
Диссоциация димеров цитохрома c
Гомодимеры цитохрома c легко образуются из растворов для электрораспыления высокой концентрации (~ 100 мкм M) с зарядовыми состояниями в диапазоне от [(цитохром c ) 2 + 19H] 19 + к [(цитохром c ) 2 + 13H] 13+ (сокращенно D19 – D13).Как было показано ранее, диссоциация цитохрома c D19, образованного непосредственно из раствора, приводит к узкому симметричному процессу разделения заряда [56]. МС / МС D17, образованного непосредственно из раствора, приводит к относительно широкому, но симметричному процессу разделения заряда (преимущественно D17 → M9 + M8) ().
МС / МС спектры ( a ) (димеры цитохрома c + 17H) 17+ , (D17), образованные непосредственно ESI, ( b ) (димеры цитохрома c + 13H) 13+ , (D13), образованный выделением D19-D17, восстановлением газофазного переноса протона диэтиламином, выделением и последующей активацией D13, и ионы ( c ) D13, образованные непосредственно из раствора.Ионы образовывались из 100 мМ раствора (вода: метанол, 1: 1 + 2% уксусная кислота).
Напротив, диссоциация D13, образованного непосредственно из раствора, приводит к сильно асимметричному распределению заряда с центром вокруг D13 → M9 + M4 (). Ионы D17-D19 были изолированы и прореагировали с триэтиламином внутри ионной ячейки FT-ICR в течение 100 с, что привело к образованию преимущественно ионов D13. Выделение D13 и последующая активация этих ионов дает спектр МС / МС, показанный на. Доминирующим процессом является симметричная диссоциация с центром D13 → M7 + M6.Таким образом, процесс диссоциации D13 явно зависит от того, образуются ли они в результате восстановления с переносом протона в газовой фазе D17 – D19 () или образуются непосредственно из раствора (). В этих экспериментальных условиях внутренняя энергия иона уравновешивается с полем излучения черного тела при комнатной температуре до активации иона [63]. Таким образом, различие во фрагментации не может быть артефактом различия внутренней энергии. Эти результаты показывают, что структуры ионов D13 различаются для этих двух методов образования ионов и что структура димера влияет на то, как эти димеры диссоциируют, т.е.е., через симметричное и асимметричное разделение заряда. Следует отметить, что димеризация в растворе неспецифична и что может присутствовать большой ансамбль димерных структур. Однако ясно, что существуют по крайней мере две отдельные структуры или различные ансамбли структур.
«Память» ионов D13 может быть связана с различными димерными структурами, существующими в растворе. Эти димеры могут получать разное количество зарядов в процессе ионизации электрораспылением, или процесс электрораспыления может вызывать конформационные изменения в некоторой части димеров при переходе из раствора в газовую фазу, что приводит к различным структурам димеров для D17 по сравнению с D13.Мы не можем четко различать эти две возможности, основываясь только на этих результатах. Однако широкое распределение в зарядовых состояниях димеров, образованных в этих денатурирующих условиях, по сравнению с типичным единственным доминирующим зарядовым состоянием димера, наблюдаемым при электрораспылении из растворов, в которых эти молекулы имеют нативную структуру, предполагает, что первый механизм более вероятен, т. Е. в растворе существуют по крайней мере две отдельные структуры или ансамбль структур, и эти структуры в конечном итоге имеют разную степень заряда в процессе электрораспыления.
Аналогичные результаты наблюдаются для ионов D15, образованных из раствора, по сравнению с ионами, образованными переносом протона. Те, которые образуются непосредственно из раствора, демонстрируют бимодальную диссоциацию: симметричный процесс, сосредоточенный вокруг M8 / M7, и асимметричный процесс, сосредоточенный вокруг M10 / M5 (). Восстановление заряда D19 – D17 с образованием более низких зарядовых состояний с последующим выделением и диссоциацией D15 приводит только к симметричной диссоциации с образованием M8 / M7 (). Опять же, эти ионы D15 явно сохраняют «память» о том, образовались ли они в газовой фазе из более высоких зарядовых состояний, которые симметрично диссоциируют, или непосредственно из раствора.
МС / МС спектры (цитохрома c димеры + 15H) 15+ , (D15), образованного ( a ) непосредственно ESI и ( b ), образованного выделением D19-D17, газ- восстановление фазового переноса протона диэтиламином, выделение и последующая активация D15. Ионы образовывались из 100 мМ раствора (вода: метанол, 1: 1 + 2% уксусная кислота).
Газофазный H / D обмен мономеров и димеров цитохрома c с ND
3Предыдущие исследования показали, что степень асимметрии заряда цитохрома c D11, образованного непосредственно наноэлектрораспылением с использованием водных растворов, содержащих 100 мМ ацетата аммония нейтральный pH зависел от внутренней энергии, которая вкладывалась в ионы [56].Чтобы попытаться установить, в какой степени различные конформеры могут вносить вклад в наблюдаемую энергетическую зависимость, на D11 были проведены исследования обмена H / D. Тщательная настройка параметров источника ESI, в частности увеличение напряжения смещения на внешнем гексаполе накопления и увеличение времени нагрузки гексаполя, способствует образованию цитохрома c D11 ().
ESI масс-спектр 500 мкМ M конский цитохром c в водном растворе со 100 мМ ацетатом аммония, pH ~ 7, со спектрометром, настроенным на получение максимального количества димеров с зарядами 11+ (D11).
Для этих экспериментов по газофазному H / D обмену давление NH 3 изменялось от 1 × 10 –8 до 3 × 10 –7 торр с постоянным временем реакции 180 с. Для эксперимента при самом высоком давлении реагента (наибольшая степень обмена H / D) () среднее количество атомов водорода, обмениваемых на дейтерий, составляет ~ 170 после вычитания обмена 11 протонов. Ширина распределения составляет примерно 110 Да. Отношение сигнал / шум недостаточно, чтобы приписать вариации интенсивностей изотопов разрешимым конформерам.Напротив, M7 в тех же условиях показывает средний обмен ~ 84 атомов водорода на дейтерий с шириной изотопического распределения ~ 50. Таким образом, как средняя степень обмена, так и ширина распределения димера примерно вдвое больше, чем у мономера. Если бы присутствовала только одна структура, нельзя было бы ожидать, что суммарная максимальная степень обмена и ширина распределения будут линейно увеличиваться с массой иона, если предположить, что степень обмена H / D связана с открытой поверхностью кластера.Эти результаты предполагают присутствие множественных конформеров димеров, которые не разрешаются в этих экспериментах по обмену H / D, что согласуется с нашим ожиданием, что эти ионы D11 образуются в результате неспецифической димеризации молекул циктохрома c в растворе.
Частичные масс-спектры ESI цитохрома c (показана область D11) при тех же условиях раствора, что и раньше, и после газофазного водородно-дейтериевого обмена с ND 3 в течение 180 с при ( b ) 1 × 10 −7 торр и ( c ) 3 × 10 −7 торр.
Обширные исследования газофазного H / D обмена мономеров цитохрома c были проведены McLafferty и соавторами [22–24], которые обнаружили целых семь конформаций цитохрома c M7, образованных непосредственно ESI [24]. В наших экспериментах в качестве обменного реагента используется ND 3 вместо D 2 O [24], и время реакции короче. Распределение изотопов для M7 предполагает наличие нескольких конформеров, согласующихся с результатами, полученными Маклафферти и соавторами.
Выводы
Асимметричное распределение заряда, которое наблюдается при диссоциации гомодимеров белков, можно объяснить двумя различными механизмами. В одном механизме асимметрия индуцируется разворачиванием одного из белков в димере при активации. В димерах, состоящих из двух почти идентичных белков, белок с более высокой конформационной гибкостью отделяется от комплекса с большим зарядом. Это может быть связано с тем, что часть молекулы покидает барьер диссоциации, где дальнейшее развертывание молекулы индуцируется отталкивающим кулоновским потенциалом.Во время этого процесса разворачивания и разделения протоны передаются разворачивающемуся белку, чтобы уменьшить общую кулоновскую энергию. Этот механизм лучше всего объясняет результаты, наблюдаемые для гетеродимеров α -лактабумина, состоящих из одного окисленного и одного восстановленного белка. Этот механизм также лучше всего объясняет результаты для димеров цитохрома c с 11 зарядами, для которых наблюдается энергетическая зависимость в степени асимметрии [56].
Некоторая часть наблюдаемого асимметричного распределения заряда также может быть приписана другому механизму, в котором структуры мономеров в димере различаются.Это лучше всего объясняет результаты, наблюдаемые для димеров цитохрома c с зарядами 17+ по сравнению с димерами с 13+ зарядами. При образовании непосредственно из раствора первый диссоциирует с симметричным распределением заряда, тогда как последний диссоциирует с сильно асимметричным распределением заряда. Однако, когда зарядовое состояние ионов димера 17+ снижается до 13+ в газовой фазе посредством мягких реакций переноса протона, образующиеся ионы 13+ диссоциируют с симметричным распределением заряда.Это ясно демонстрирует, что ионы 13+, образованные из ионов 17+, имеют другую структуру (или разные ансамбли структур), чем те, которые образуются непосредственно из раствора, и что это структурное различие отвечает за асимметричное и симметричное распределение заряда, наблюдаемое при диссоциации этих 13+. ионы. Это первое зарегистрированное свидетельство того, что асимметричное распределение заряда может быть вызвано структурными различиями в субъединицах мономера в комплексе.
Наконец, H / D обмен зарядового состояния 11+ димеров цитохрома c указывает на присутствие множественных конформеров, хотя эти конформеры не разрешаются в этом эксперименте.Дальнейшая работа с другими реагентами для обмена H / D или в других условиях, или с конформационно-селективными методами, такими как подвижность ионов, в сочетании с экспериментами по диссоциации может предоставить дополнительную полезную информацию о процессе разделения заряда. Лучшее понимание этого процесса в конечном итоге улучшит структурную информацию, которая может быть получена при газофазной диссоциации нековалентных комплексов.
Благодарности
Авторы благодарят доктора Дэвида Кинга (Калифорнийский университет, Беркли) за восстановление и очистку α -лактальбумина.Эта работа была поддержана Национальным институтом здравоохранения (грант № R01-GM64712-01).
Ссылки
1. Калета Д.Т., Джарролд М.Ф. Пептидные вертушки. J Am Chem Soc. 2002; 124: 1154–1155. [PubMed] [Google Scholar] 2. Kaleta DT, Jarrold MF. Нековалентные взаимодействия между несольватированными пептидами. J. Phys Chem A. 2002; 106: 9655–9664. [Google Scholar] 3. Clemmer DE, Jarrold MF. Измерения подвижности ионов и их приложения к кластерам и биомолекулам. J. Mass Spectrom. 1997. 32: 577–592. [Google Scholar] 4.Counterman AE, Valentine SJ, Srebalus CA, Henderson SC, Hoaglund CS, Clemmer DE. Структура высокого порядка и диссоциация газообразных пептидных агрегатов, скрытых в масс-спектрах. J Am Soc масс-спектрометрия. 1998. 9: 743–759. [PubMed] [Google Scholar] 5. Counterman AE, Hilderbrand AE, Barnes CAS, Clemmer DE. Формирование пептидных агрегатов во время ESI: размер, заряд, состав и вклад в шум. J Am Soc масс-спектрометрия. 2001; 12: 1020–1035. [Google Scholar] 6. Гидден Дж., Виттенбах Т., Батька Дж. Дж., Вайс П., Джексон А. Т., Скривенс Дж. Х., Бауэрс МТ.Энергетика сворачивания и динамика макромолекул в газовой фазе: щелочные ионно-катионизированные олигомеры поли (этилентерефталата). J Am Chem Soc. 1999; 121: 1421–1422. [Google Scholar] 7. Гевремонт Р., Purves RW. Спектрометрия-масс-спектрометрия подвижности ионов с асимметричной формой волны в высоком поле: исследование ионов лейцина-энкефалина, полученных с помощью ионизации электрораспылением. J Am Soc масс-спектрометрия. 1999; 10: 492–501. [PubMed] [Google Scholar] 8. Purves RW, Barnett DA, Guevremont R. Разделение белков-конформеров с использованием спектрометрии-масс-спектрометрии асимметричной формы волны с асимметричной формой волны электроспрея.Int J масс-спектрометрия. 2000; 197: 163–177. [Google Scholar] 9. Дэн Ю.З., Чжан З.К., Смит Д.Л. Сравнение непрерывного и импульсного мечения амидного водородного обмена / масс-спектрометрии для исследования динамики белков. J Am Soc масс-спектрометрия. 1999; 10: 675–684. [PubMed] [Google Scholar] 10. Чжу М.М., Ремпель Д.Л., Брутто М.Л. Данные моделирования титрования, амидного H / D обмена и масс-спектрометрии для получения констант связывания белок-лиганд. J Am Soc масс-спектрометрия. 2004. 15: 388–397. [PubMed] [Google Scholar] 11. Гарда Э., Уилларда Д., Брегара Дж., Грин МК, Лебрилла CB.Сайт-специфичность в H-D-обменных реакциях газофазных протонированных аминокислот с Ch4OD. Org Mass Spectrom. 1993; 28: 1632–1639. [Google Scholar] 12. Кэмпбелл С., Роджерс М. Т., Марцлафф Е. М., Бошам Дж. Л.. Структурные и энергетические ограничения на реакции газофазного водородно-дейтериевого обмена протонированных пептидов с D2O, CD3OD, CD3CO2D и ND3. J Am Chem Soc. 1994; 116: 9765–9766. [Google Scholar] 13. Gard E, Green MK, Bregar J, Lebrilla CB. Газофазный водород-дейтериевый обмен как молекулярный зонд для взаимодействия метанола и протонированных пептидов.J Am Soc масс-спектрометрия. 1994; 5: 623–631. [PubMed] [Google Scholar] 14. Кэмпбелл С., Роджерс М. Т., Марцлафф Е. М., Бошам Дж. Л.. Реакции обмена дейтерия как зонд структуры биомолекул. Фундаментальные исследования газофазных реакций H / D обмена протонированных олигомеров глицина с D2O, CD3OD, CD3CO2D и ND3. J Am Chem Soc. 1995; 117: 12840–12854. [Google Scholar] 15. Грин МК, Penn SG, Lebrilla CB. Комплексообразование протонированных пептидов с сахаридами в газовой фазе снижает скорость реакций обмена водород / дейтерий.J Am Soc масс-спектрометрия. 1995; 6: 1247–1251. [PubMed] [Google Scholar] 16. Калташов И.А., Дорошенко В.М., Коттер Р.Дж. Газофазные реакции обмена пептидных ионов водород / дейтерий в квадрупольном масс-спектрометре с ионной ловушкой. Белки. 1997. 28: 53–58. [PubMed] [Google Scholar] 17. Heck AJR, Jorgensen TJD, O’Sullivan M, von Raumer M, Derrick PJ. Газофазные нековалентные взаимодействия между антибиотиками группы ванкомицина и пептидами-предшественниками бактериальной клеточной стенки, вызванные обменом водорода / дейтерия. J Am Soc масс-спектрометрия.1998. 9: 1255–1266. [Google Scholar] 18. Фрейтас М.А., Маршалл АГ. Скорость и степень газофазного водородно-дейтериевого обмена брадикининов: данные о пептидных цвиттерионах в газовой фазе. Int J масс-спектрометрия. 1999. 183: 221–231. [Google Scholar] 19. Рейзер ML, Brodbelt JS. Газофазные реакции обмена H / D полиаминовых комплексов: [M + H) (+), (M плюс щелочной металл (+)] и (M + 2H) (2+) J Am Soc Mass Spectrom. 2000; 11 : 711–721. [PubMed] [Google Scholar] 20. Леви-Сери Э, Костер Г., Коган А., Гутман К., Рубен Б. Г., Лифшиц К.Исследование H / D-обмена в протонированном брадикинине с помощью ионизационной трубки с электрораспылением. J. Phys Chem A. 2001; 105: 5552–5559. [Google Scholar] 21. Солоуки Т., Форт Р.С., Аломари А., Фаттахи А. Реакции газофазного водородно-дейтериевого обмена модельного пептида: FT-ICR и вычислительный анализ конформационных мутаций, индуцированных металлами. J Am Soc масс-спектрометрия. 2001; 12: 1272–1285. [PubMed] [Google Scholar] 22. Suckau D, Shi Y, Beu SC, Senko MW, Quinn JP, Wampler FM, McLafferty FW. Сосуществующие стабильные конформации газообразных белковых ионов.Proc Nat Acad Sci USA. 1993; 90: 790–793. [Бесплатная статья PMC] [PubMed] [Google Scholar] 23. Wood TD, Chorush RA, Wampler FM, Little DP, O’Connor PB, McLafferty FW. Газофазное сворачивание и разворачивание катионов цитохрома- c . Proc Natl Acad Sci USA. 1995; 92: 2451–2454. [Бесплатная статья PMC] [PubMed] [Google Scholar] 24. McLafferty FW, Guan ZQ, Haupts U, Wood TD, Kelleher NL. Газообразные конформационные структуры цитохрома c. J Am Chem Soc. 1998. 120: 4732–4740. [Google Scholar] 25. Рид Г.Е., О’Хэр RAJ, Styles ML, McFadyen WD, Simpson RJ.Газофазные ионно-молекулярные реакции в модифицированной ионной ловушке: H / D-обмен нековалентных комплексов и координационно-ненасыщенных комплексов платины. Масс-спектрометр Rapid Commun. 1998; 12: 1701–1708. [Google Scholar] 26. Коган А., Устюжанин П., Рубен Б. Г., Лифшиц С. Водород / дейтериевый обмен мономеров и димеров лейцин-энкефалина. Int J масс-спектрометрия. 2002; 213: 1–4. [Google Scholar] 27. Мао Д., Дуглас ди-джей. H / D-обмен газофазными ионами брадикинина в линейной квадрупольной ионной ловушке. J Am Soc масс-спектрометрия.2003. 14: 85–94. [PubMed] [Google Scholar] 28. Ли SW, Ли Х.Н., Ким Х.С., Бошам Дж. Л.. Селективное связывание краун-эфиров с протонированными пептидами может быть использовано для исследования механизмов обмена H / D и реакций диссоциации, вызванных столкновениями, в газовой фазе. J Am Chem Soc. 1998. 120: 5800–5805. [Google Scholar] 29. Валовой DS, Schnier PD, Rodriguezcruz SE, Fagerquist CK, Williams ER. Конформации и сворачивание ионов лизоцима в вакууме. Proc Nat Acad Sci USA. 1996; 93: 3143–3148. [Бесплатная статья PMC] [PubMed] [Google Scholar] 31.Loo JA. Изучение нековалентных белковых комплексов методом масс-спектрометрии с ионизацией электрораспылением. Масс-спектром. Ред. 1997; 16: 1–23. [PubMed] [Google Scholar] 32. Лим Х.К., Сие Й.Л., Ганем Б., Хенион Дж. Распознавание пептидных лигандов клеточной стенки антибиотиками группы ванкомицина – исследования с использованием масс-спектрометрии с ионным распылением. J. Mass Spectrom. 1995; 30: 708–714. [Google Scholar] 33. Лоо Дж. А., Ху П. Ф., МакКоннелл П., Мюллер В. Т., Сойер Т. К., Танабал В. Исследование взаимодействий связывания белка с фосфопептидом домена Src Sh3 с помощью масс-спектрометрии с ионизацией электрораспылением.J Am Soc масс-спектрометрия. 1997. 8: 234–243. [Google Scholar] 34. Йоргенсен TJD, Roepstorff P, Heck AJR. Прямое определение констант связывания раствора для нековалентных комплексов между пептидными аналогами бактериальной клеточной стенки и антибиотиками группы ванкомицина методом масс-спектрометрии с ионизацией электрораспылением. Anal Chem. 1998; 70: 4427–4432. [Google Scholar] 35. Дэниел Дж. М., Фрисс С. Д., Раджагопалан С., Вендт С., Зеноби Р. Количественное определение нековалентных связывающих взаимодействий с использованием масс-спектрометрии с мягкой ионизацией.Int J масс-спектрометрия. 2002; 216: 1-27. [Google Scholar] 36. Валовой DS, Чжао YX, Уильямс ER. Диссоциация комплексов гем-глобин с помощью инфракрасной радиационной диссоциации черного тела: молекулярная специфичность в газовой фазе? J Am Soc масс-спектрометрия. 1997. 8: 519–524. [Бесплатная статья PMC] [PubMed] [Google Scholar] 37. Schnier PD, Klassen JS, Strittmatter EE, Williams ER. Энергии активации для диссоциации двухцепочечных олигонуклеотидных анионов: данные о спаривании оснований Уотсона-Крика в вакууме. J Am Chem Soc. 1998; 120: 9605–9613.[Бесплатная статья PMC] [PubMed] [Google Scholar] 38. Рогалевич Ф., Хоппиллиард Я., Оханесян Г. Структуры и фрагментация комплексов цинка (II) аминокислот в газовой фазе. IV. Влияние растворителя на структуру электрораспыленных ионов. Int J масс-спектрометрия. 2003. 227: 439–451. [Google Scholar] 39. Габелика В., Де Пау Э. Сравнение между фазовой стабильностью в растворе и кинетической стабильностью в газовой фазе олигодезоксинуклеотидных дуплексов. J. Mass Spectrom. 2001; 36: 397–402. [PubMed] [Google Scholar] 40. Габелика V, Де Пау Э.Сравнение индуцированной столкновением диссоциации дуплексной ДНК при различных режимах коллизии: доказательства многоступенчатого механизма диссоциации. J Am Soc масс-спектрометрия. 2002; 13: 91–98. [PubMed] [Google Scholar] 41. Габелика В., Де Пау Э. Диссоциация 16-мерных дуплексов ДНК с различными последовательностями, вызванная столкновением: свидетельства сохранения конформации двойной спирали в газовой фазе. Int J масс-спектрометрия. 2002; 219: 151–159. [Google Scholar] 42. Ван К.Х., Шибуэ Т., Брутто ML. Нековалентные комплексы между ДНК-связывающими лекарствами и двухцепочечными олигодезоксинуклеотидами: исследование методом масс-спектрометрии с ионной ловушкой ESI.J Am Chem Soc. 2000; 122: 300–307. [Google Scholar] 43. Чен Ю.Л., Кэмпбелл Дж. М., Коллингс Б. А., Конерманн Л., Дуглас Д. Д.. Устойчивость высокозаряженного нековалентного комплекса в газовой фазе: голомиоглобина. Масс-спектрометр Rapid Commun. 1998. 12: 1003–1010. [PubMed] [Google Scholar] 44. Хантер С.Л., Маук А.Г., Дуглас Диджей. Диссоциация гема от миоглобина и цитохрома b (5): сравнение поведения в растворе и газовой фазе. Биохим США. 1997; 36: 1018–1025. [PubMed] [Google Scholar] 45. Йоргенсен Т.Д., Делфорж Д., Ремакл Дж., Бойсен Дж., Рёпсторф П.Индуцированная столкновением диссоциация нековалентных комплексов между антибиотиками ванкомицина и стереоизомерами пептидного лиганда: доказательства молекулярного распознавания в газовой фазе. Int J масс-спектрометрия. 1999. 188: 63–85. [Google Scholar] 46. Loo JA, He JX, Cody WL. Структура высшего порядка в газовой фазе отражает структуру раствора. J Am Chem Soc. 1998; 120: 4542–4543. [Google Scholar] 47. Ростом А.А., Фучини П., Бенджамин Д.Р., Юенеманн Р., Нирхаус К.Х., Хартл Ф.У., Добсон С.М., Робинсон К.В. Обнаружение и селективная диссоциация интактных рибосом в масс-спектрометре.Proc Nat Acad Sci USA. 2000; 97: 5185–5190. [Бесплатная статья PMC] [PubMed] [Google Scholar] 48. Pinkse MWH, Maier CS, Kim JI, Oh BH, Heck AJR. Макромолекулярная оценка уреазы Helicobacter pylori, исследованная методом масс-спектрометрии. J. Mass Spectrom. 2003. 38: 315–320. [PubMed] [Google Scholar] 49. Лайт-Вал К.Дж., Шварц Б.Л., Смит Р.Д. Наблюдение нековалентных четвертичных ассоциаций белков с помощью масс-спектрометрии с ионизацией электрораспылением. J Am Chem Soc. 1994; 116: 5271–5278. [Google Scholar] 50. Шварц Б.Л., Брюс Дж. Э., Андерсон Г. А., Хофстадлер С. А., Роквуд А. Л., Смит Р. Д., Чилкоти А., Стейтон П. С..Диссоциация тетрамерных ионов нековалентных стрептавидиновых комплексов, образующихся при ионизации электрораспылением. J Am Soc масс-спектрометрия. 1995; 6: 459–465. [PubMed] [Google Scholar] 51. Фицджеральд М.С., Чернушевич И., Стэндинг К.Г., Уитмен С.П., Кент SBH. Исследование олигомерной структуры фермента методом времяпролетной масс-спектрометрии с ионизацией электрораспылением. Proc Nat Acad Sci USA. 1996; 93: 6851–6856. [Бесплатная статья PMC] [PubMed] [Google Scholar] 52. Фелицын Н., Китова Е.Н., Классен Я.С. Термическое разложение газообразного мультибелкового комплекса, исследованное методом черной инфракрасной радиационной диссоциации.Исследование происхождения асимметричного поведения диссоциации. Anal Chem. 2001; 73: 4647–4661. [PubMed] [Google Scholar] 53. Бенеш JLP, Соботт Ф., Робинсон CV. Термическая диссоциация мультимерных белковых комплексов с помощью масс-спектрометрии с наноэлектрораспылением. Anal Chem. 2003. 75: 2208–2214. [PubMed] [Google Scholar] 55. Versluis C, van der Staaij A, Stokvis E, Heck AJR, de Craene B. Образование метастабильных ионов и разделение разрозненных зарядов при газофазной диссекции белковых сборок, изученных методом ортогональной времяпролетной масс-спектрометрии.J Am Soc масс-спектрометрия. 2001; 12: 329–336. [PubMed] [Google Scholar] 56. Jurchen JC, Williams ER. Происхождение асимметричного разделения заряда при диссоциации газофазных белковых гомодимеров. J Am Chem Soc. 2003; 125: 2817–2826. [Бесплатная статья PMC] [PubMed] [Google Scholar] 57. Jurchen JC, Garcia DE, Williams ER. Пути газофазной диссоциации многозарядных пептидных кластеров. J Am Soc масс-спектрометрия. 2003. 14: 1373–1386. [Бесплатная статья PMC] [PubMed] [Google Scholar] 58. Готье Дж. В., Траутман Т. Р., Якобсон Д.Б.Продолжительное нерезонансное облучение для диссоциации, активируемой столкновениями, с использованием метода масс-спектрометрии с преобразованием Фурье и диссоциации, активируемой столкновениями, которая имитирует инфракрасную многофотонную диссоциацию. Анальный Чим Акта. 1991; 246: 211–225. [Google Scholar] 59. Вингер Б.Э., Лайт-Валь КДЖ, Роквуд А.Л., Смит Р.Д. Исследование качественных различий конформации многократно протонированных газофазных белков с помощью изотопного обмена H / D с D2O. J Am Chem Soc. 1992; 114: 5897–5898. [Google Scholar] 60. Фелицын Н., Китова Е.Н., Классен Я.С.Термическая диссоциация гомодимера белка экотина в газовой фазе. J Am Soc масс-спектрометрия. 2002; 13: 1432–1442. [PubMed] [Google Scholar] 61. Апостол I. Оценка относительной стабильности сконструированных гемоглобинов с помощью масс-спектрометрии с электрораспылением. Анальная биохимия. 1999; 272: 8–18. [PubMed] [Google Scholar] 62. Versluis C, Heck AJR. Газофазная диссоциация гемоглобина. Int J масс-спектрометрия. 2001; 210: 637–649. [Google Scholar] 63. Цена WD, Williams ER. Активация пептидных ионов излучением черного тела: факторы, приводящие к кинетике диссоциации в пределе быстрого обмена энергией.J. Phys Chem A. 1997; 101: 8844–8852. [Бесплатная статья PMC] [PubMed] [Google Scholar]Дальнейшие исследования происхождения асимметричного разделения заряда в белковых гомодимерах
J Am Soc Mass Spectrom. Авторская рукопись; доступно в PMC 2006 6 января.
Опубликован в окончательной редакции как:
PMCID: PMC1325214
NIHMSID: NIHMS3986
Химический факультет Калифорнийского университета в Беркли, Беркли, Калифорния, США
Запросы на перепечатку адресов: ДокторЭ. Р. Уильямс, химический факультет Калифорнийского университета в Беркли, Беркли, Калифорния 94720, США. Электронное письмо: ude.yelekreb.mehcc@smailliw См. другие статьи в PMC, в которых цитируется опубликованная статья.Abstract
Диссоциация протонированных в газовой фазе белковых димеров на составляющие их мономеры может приводить к симметричному или асимметричному распределению заряда. Диссоциация гомодимеров α -лактальбумина с 15+ зарядами приводит к симметричному, но широкому распределению белковых мономеров с зарядовыми состояниями, сосредоточенными вокруг 8 + / 7 +.Напротив, диссоциация гетеродимера 15+, состоящего из одной молекулы в окисленной форме и одной в восстановленной форме, приводит к сильно асимметричному распределению зарядов, при котором восстановленные частицы уносят преимущественно заряды 11+, а окисленная молекула уносит заряды 4+. . Этот результат не может быть адекватно объяснен дифференциальной зарядкой, происходящей ни в растворе, ни в процессе электрораспыления, но, по-видимому, лучше всего объясняется восстановленными частицами, разворачивающимися при активации в газовой фазе с последующим разделением и переносом протонов на разворачивающиеся частицы в диссоциативном комплексе. чтобы минимизировать кулоновское отталкивание.Для димеров цитохрома c , образованных непосредственно из раствора, зарядовое состояние 17+ подвергается симметричному распределению заряда, тогда как диссоциация 13+ асимметрична. Уменьшение зарядового состояния димеров с зарядами 17+ до 13+ посредством переноса протона в газовой фазе и последующей диссоциации выбранных по массе ионов 13+ приводит к симметричному распределению заряда. Этот результат ясно показывает, что структура димерных ионов с зарядами 13+ зависит от метода образования иона и что структурные различия ответственны за наблюдаемое симметричное и асимметричное разделение зарядов.Это указывает на то, что асимметрия, наблюдаемая, когда эти ионы образуются непосредственно из раствора, должна происходить либо из-за различий в конформациях мономера в димере, которые существуют в растворе, либо в процессе ионизации электрораспылением. Эти результаты предоставляют дополнительные доказательства происхождения асимметрии заряда, которая возникает при диссоциации многозарядных белковых комплексов, и показывают, что некоторая информация о фазе раствора может быть получена из этих экспериментов по газофазной диссоциации.
Биологическая функциональность белков и белковых комплексов сильно зависит от их структур в фазе раствора. Важность области структурной биологии отражена в нескольких недавно присужденных Нобелевских премиях за вклад в методы структурного выяснения, включая ЯМР (Курт Вутрих, 2002 г. по химии), рентгеновскую кристаллографию (Иоганн Дайзенхофер, Роберт Хубер и Хартмут Мишель, 1988 г. в. химия; Герберт Хауптман и Джером Карл 1985 по химии) и кристаллографическая электронная микроскопия (сэр Аарон Клаг 1982 по химии).Анализ белков и белковых комплексов этими методами может предоставить подробную трехмерную информацию, но эти методы часто ограничены экспериментальными ограничениями, такими как необходимость получения полезных кристаллов белка или требование значительного количества образца. Напротив, для масс-спектрометрии требуется небольшой образец, а с ионизацией электрораспылением (ESI) или мягкой лазерной десорбцией / ионизацией (John Fenn and Kiochi Tanaka, 2002 по химии) в масс-спектрометр можно ввести большое количество биомолекул и специфических комплексов.Был разработан ряд методов для исследования общих трехмерных форм ионов биомолекул и нековалентных комплексов с использованием методов ионной подвижности [1–8], H / D обмена как в растворе [9, 10], так и в газовой фазе [ 11–28] и реакционной способности с переносом протона [29, 30].
Для анализа нековалентных комплексов масс-спектрометрия имеет то преимущество, что стехиометрию комплекса можно определить с помощью простого измерения массы [31]. В некоторых случаях MS может обеспечить правильные константы связывания в фазе раствора для белок-лигандных взаимодействий [32–35].Эти разработки подробно рассмотрены [31, 35]. В принципе, информацию о структуре нековалентных комплексов можно потенциально получить, исследуя ионы продуктов, образующиеся в результате диссоциации комплекса, в эксперименте тандемной масс-спектрометрии (МС / МС). Объем соответствующей информации, которую можно получить в результате этих экспериментов, зависит от количества структурной информации о фазе раствора, которая сохраняется биологическими комплексами при переходе в газовую фазу (или от того, может ли эта информация быть восстановлена на основе сведений о происходящих структурных изменениях. во время этого перехода) и степень, в которой методы MS / MS нарушают любую оставшуюся структурную информацию.Многие эксперименты показывают, что газофазные комплексы биомолекул и даже некоторые небольшие молекулы могут сохранять «память» о своей фазовой структуре раствора, и что эта память может быть исследована процессами диссоциации [36–48].
Ряд исследований гомогенных биологических комплексов (тетрамеры, пентамеры, гексамеры и т. Д.) С помощью МС / МС выявили первоначально неожиданный результат, заключающийся в том, что первичный путь диссоциации мультимерных комплексов представляет собой выброс высоко заряженного мономера, который удаляет 30-50 % заряда комплекса [49–53].Рентгеновская кристаллография показывает, что структуры растворов субъединиц во многих гомогенных комплексах идентичны. В некоторых случаях весь комплекс кристаллизуется внутри асимметричной единицы кристалла, как в случае с шига-подобным токсином [9], в то время как в других, таких как апострептавидин, асимметричная единица содержит одну субъединицу, и структура комплекс экстраполирован с помощью молекулярного моделирования [54]. Из-за практически идентичных структур фазы раствора участвующих субъединиц в этих биологических комплексах наблюдаемое асимметричное распределение заряда, по-видимому, является артефактом процедуры ESI-MS / MS.Было высказано предположение, что асимметричное распределение заряда коррелирует с газофазными конформационными перестройками [50, 52, 55, 56]. Также было продемонстрировано, что степень асимметричного распределения заряда для гомодимеров белков зависит от конформационной гибкости белков в комплексе, зарядового состояния комплекса, отложения внутренней энергии и состава растворов для электрораспыления, из которых комплексы образуются. формируются [56]. Для более крупных кластеров, состоящих из идентичных субъединиц, наблюдалось деление с преимущественным образованием протонированных димеров, а с увеличением зарядового состояния и размера кластера наблюдалось усиление образования протонированного мономера [57].Степень деления по сравнению с потерями нейтралов для этих кластеров может быть связана с наблюдаемой для многозарядных металлических кластеров с одним важным отличием: различия в конформационной гибкости субъединиц мономера внутри кластера приводят к различиям в коэффициентах ветвления различных диссоциаций. процессы, наблюдаемые для этих многократно протонированных кластеров биомолекул [57].
Газофазный водород-дейтериевый (H / D) обмен обычно осуществляется путем хранения интересующего иона в вакууме и измерения изменения массы иона из-за реакций обмена H / D, которые происходят во время столкновений с нейтральными молекулами обменного реагента. , например, D 2 O или ND 3 .Хотя газофазный обмен H / D широко использовался для характеристики пептидов и малых молекул [11–21], а также структуры некоторых газофазных белков [22–24], очень мало исследований было выполнено с нековалентными комплексами, включающими биологические ионы, и эта нековалентная работа была сосредоточена на димерах пептидов [25, 26] и пептидных комплексах с ионами металлов [16, 18, 21, 27] и небольшими молекулами [15, 28].
Здесь представлена дальнейшая характеристика гомодимеров белка с использованием газофазной диссоциации и газофазного H / D обмена.Исследования диссоциации смешанных димеров окисленного и восстановленного α -лактальбумина предоставляют дополнительные доказательства того, что конформационная гибкость играет ключевую роль в определении степени асимметричного распределения заряда. Для диссоциации гомодимеров цитохрома c показано, что зарядовые состояния, полученные в результате снижения заряда в газовой фазе из гомодимеров с более высоким зарядовым состоянием, диссоциируют совсем иначе, чем состояния, полученные непосредственно из раствора. Представлена первая демонстрация газофазного H / D обмена белкового комплекса.Эти результаты показывают, что два разных механизма ответственны за асимметричное распределение заряда, наблюдаемое при диссоциации гомодимеров белка.
Experimental
Эксперименты проводились на масс-спектрометре Berkeley-Bruker с узким проходом 9,4 Тл с ионно-циклотронным резонансом с преобразованием Фурье (FT-ICR), который описан в другом месте [56]. Используемые экспериментальные процедуры аналогичны использованным ранее [56]. Краткое описание и дополнительные сведения приведены ниже.
Chemicals
Цитохром сердца лошади c , α -лактальбумин из коровьего молока и триэтиламин были приобретены у Sigma-Aldrich Co. (Сент-Луис, Миссури) и использовались без дополнительной очистки. Дейтерированный аммиак был приобретен в Cambridge Isotope Laboratories (Woburn, MA). Ацетамидирование α -лактальбумина было выполнено доктором Дэвидом Кингом (Калифорнийский университет, Беркли). Все четыре внутримолекулярные дисульфидные связи α -лактальбумина восстанавливаются трис (карбоксиэтил) фосфином, ацетамидируются в темноте йодацетамидом в течение одного часа и очищаются с помощью ВЭЖХ для удаления буфера и непрореагировавших реагентов.Практически полное восстановление и ацетамидирование всех дисульфидных связей в α -лактальбумине было подтверждено измерениями массы.
Образование димеров
Образование неспецифических белковых димеров усиливается за счет использования концентраций белка 100–500 мкМ M и регулирования напряжений на границе раздела для «мягкого» введения ионов. Сигнал димера улучшается за счет настройки на селективное накопление ионов во внешнем гексаполе в области источника прибора. Это достигается накоплением ионов в гексаполе в течение 3–4 с (против ≤ 1.0 с), увеличивая смещение постоянного тока гексаполя с ~ 2,7 В до 3,5–4,5 В и вводя несколько скоплений гексаполя в ионную ячейку перед обнаружением.
Смешанные димеры окисленного (все четыре дисульфидные связи не повреждены) и восстановленного (все четыре дисульфидные связи разорваны и все восемь остатков цистеина впоследствии ацетамидированы) α -лактальбумин были образованы путем смешивания растворов двух форм α -лактальбумина в различных соотношения. Концентрация общего белка поддерживалась постоянной на уровне ~ 200 мкМ М, и отношения двух компонентов были изменены, чтобы максимизировать абсолютное содержание смешанного димера.Наибольшее количество смешанного димера появляется при относительной концентрации восстановленного до окисленного α -лактальбумина ~ 2: 1. Поскольку концентрации белка определялись массой лиофилизированных порошков, используемых для приготовления раствора, эти концентрации являются приблизительными. Ионы димера белка формировали с использованием ESI со скоростью потока ~ 1 мкМ л / мин в условиях раствора 1: 1 вода: метанол + 2% уксусная кислота.
Димеры цитохрома c были сформированы с использованием обычного ESI при скорости потока 1-2 мкМ л / мин и концентрации 100 мкМ М в 1: 1 вода: метанол + 2% уксусная кислота.Зарядовые состояния димеров, сформированные непосредственно из раствора, варьируются от [(цитохром c ) 2 + 19H] 19+ до [(циктохром c ) 2 + 13H] 13+ (сокращенно D19 – D13) . Зарядовые состояния димера с m / z выше, чем у D17, были выброшены из ионной ячейки с использованием коррелированных выстрелов, оставляя зарядовые состояния D19 – D17, оставшиеся в ионной ячейке. Вакуумная камера FT-ICR была подготовлена для снижения заряда путем кратковременного введения триэтиламина в вакуумную камеру под давлением ~ 3 × 10 90 · 10 7 -8 90 · 108 торр с использованием пьезоэлектрического импульсного клапана.Импульсный клапан затем закрывался через 5 с, и базовое давление прибора возвращалось к ~ 8 × 10 90 · 107 -9 90 · 108 торр через 95 с. Снижение заряда посредством переноса протона осуществлялось путем улавливания гомодимеров цитохрома c с зарядовыми состояниями D19 – D17 в приборе до тех пор, пока столкновения с остаточными молекулами триэтиламина не снижали зарядовое состояние димеров в течение 100 с, так что 13+ было самая распространенная форма димера. Вновь образованные ионы D13 были изолированы с использованием коррелированных свип-сигналов и диссоциированы с помощью длительной диссоциации, активируемой столкновениями (SORI-CAD) [58] вне резонанса и вне резонанса, при +600 Гц вне резонанса и 3.5 В пик – пик в течение 0,25 с после подачи импульса азота, повышающего давление в вакуумной камере до ~ 10 –6 торр.
Газофазный H / D обмен белковых комплексов
Газофазный H / D обмен гомодимеров цитохрома c осуществляли путем улавливания всех генерируемых электрораспылением ионов в ячейке и введения ND 3 в вакуумную камеру с использованием ручной герметичный клапан (Varian, Lexington, MA) на 180 с при нескольких давлениях в диапазоне от ~ 1 × 10 −8 до максимального ~ 3 × 10 −7 торр.После 180 с замены клапан утечки был закрыт, и вакуумная камера откачивалась в течение 60–120 с, пока базовое давление не стало достаточно низким для изотопного разрешения (<1 × 10 –8 торр). Предыдущие эксперименты по газофазному обмену H / D с белками использовали D 2 O в качестве обменного реагента [22–24, 59]. Из-за трудностей, связанных с длительным временем улавливания (> 300 с) и значительным временем, необходимым для удаления относительно нелетучего D 2 O, в качестве обменного реагента был выбран ND 3 .Давление вводимого в вакуумную камеру НД 3 измерялось некалиброванным ионным манометром, расположенным на расстоянии ~ 1,1 м от ионной ячейки.
Результаты и обсуждение
Диссоциация смешанных
димеров α -лактальбуминаИспользуя растворы ESI с высокими концентрациями белка, можно образовать как гомодимеры окисленного белка α -лактальбумина (каждый белок имеет четыре интактных дисульфидных связи), так и окисленно-восстановленные гетеродимеры α -лактальбумина (один белок имеет четыре интактных дисульфидных связи, а другой – четыре восстановленные дисульфидные связи, причем все восемь остатков цистеина ацетамидированы).И окисленные гомодимеры, и окисленно-восстановленные гетеродимеры образуются непосредственно из ESI с 15 присоединенными протонами ((Ox-Ox) 15 и (Ox-Red) 15, соответственно) (). (Ox-Ox) 15 диссоциирует с довольно широким, но симметричным распределением заряда (), аналогичным тому, что наблюдалось ранее для (Ox-Ox) 13 и (Red-Red) 17 [56]. (Red-Red) 13, как было ранее показано, асимметрично диссоциирует с преимущественным образованием M9 / M4 [56]. В отличие от (Ox-Ox) 15, (Ox-Red) 15 диссоциирует с сильно асимметричным распределением заряда ().Наиболее распространенным процессом диссоциации димера является (Ox-Red) 15 → Red11 + Ox4, процесс разделения, при котором восстановленный α -лактальбумин удаляет почти всего заряда! Обратите внимание, что большая интенсивность сигнала Red11 по сравнению с Ox4 обусловлена как более низким уровнем заряда последнего (сигнал FT-ICR пропорционален заряду), так и несколько меньшей эффективностью обнаружения при более высоком m / z .
( a ) Масс-спектр ESI, полученный из 200 мкМ раствора M (общая концентрация белка), содержащего окисленный (нетронутый дисульфид) и восстановленный α -лактальбумин (1: 1 вода: метанол + 2% уксусная кислота) , ( b ) МС / МС спектр окисленного-восстановленного гетеродимера α -лактальбумина с 15 протонами, (Ox-Red) 15 и ( c ) МС / МС спектр окисленного α -лактальбумина -окисленный гомодимер (Ox-Ox) 15, образованный из раствора ~ 125 мкМ М окисленного α -лактальбумина в 1: 1 вода: метанол + 2% уксусная кислота.Red8 представляет (восстановленный α -лактальбумин + 8H) 8+ и т. Д.
Происхождение сильно асимметричного распределения заряда может быть связано с асимметричным разделением, которое происходит в газовой фазе [56], или с асимметрией конформации или заряд мономеров, составляющих димер, либо в растворе, либо индуцированный процессами электрораспыления. В первом механизме асимметричное распределение заряда происходит из-за конформационного изменения одной из субъединиц мономера, которая составляет димер, после активации.Когда мономер разворачивается, молекула преодолевает активационный барьер для разворачивания и разделения. Из-за кулоновской энергии выход из этого барьера является сильно отталкивающим, и когда одна часть разворачивающейся молекулы выходит из этого канала, она будет управлять разворачиванием остальной части молекулы, когда она отделяется от другой субъединицы мономера. В этом процессе протоны будут переходить к выходящему разворачивающемуся иону, чтобы минимизировать кулоновскую энергию [30]. Поскольку восстановленный белок более гибок конформационно и может разворачиваться в значительно большей степени, чем окисленный белок, восстановленный белок разворачивается и уносит больший заряд, чем окисленная форма.
Альтернативный механизм этой асимметрии заряда заключается в том, что (Ox-Red) 15 может образовываться в растворе путем агрегации сильно заряженных восстановленных и низкозарядных окисленных мономеров α -лактальбумина в растворе или образовываться во время электрораспыления за счет асимметричной зарядки. отдельных белковых составляющих на заключительных этапах ESI [60]. Также было высказано предположение, что асимметричное распределение заряда для гетеродимеров может быть связано с различиями в pI [61, 62]. Хотя возникает разница в зарядке этих двух молекул, она не учитывает величину асимметричного разделения заряда.В масс-спектре ESI смешанного раствора α -лактальбумина восстановленный α -лактальбумин появляется преимущественно при 8+ и в меньшей степени при 9+ и 10+ (). Наблюдается очень мало 11+. Окисленный α -лактальбумин встречается только в зарядовом состоянии 8+. В спектре диссоциации (Ox-Red) 15 доминирующими наблюдаемыми видами являются Red11 и Ox5, несмотря на то, что численность этих видов, продуцируемых непосредственно ESI, незначительна. Хотя можно было бы рационализировать более низкое зарядовое состояние, наблюдаемое для Ox, как немного более низкое зарядовое состояние димера по сравнению с суммой зарядовых состояний отдельных частиц, это не может быть верным для восстановленных частиц, которые имеют значительно более высокий заряд при диссоциации. спектра, чем при формировании непосредственно электрораспылением.Таким образом, этот результат трудно рационализировать, основываясь только на модели, в которой асимметричное распределение заряда является либо реликтом агрегации фазы раствора, либо артефактом процесса электрораспыления. Результаты гетеродимеров (Ox-Red) 15 α -лактальбумина, по-видимому, лучше всего подтверждают процесс асимметричного распределения заряда в газовой фазе [56].
Диссоциация димеров цитохрома c
Гомодимеры цитохрома c легко образуются из растворов для электрораспыления высокой концентрации (~ 100 мкм M) с зарядовыми состояниями в диапазоне от [(цитохром c ) 2 + 19H] 19 + к [(цитохром c ) 2 + 13H] 13+ (сокращенно D19 – D13).Как было показано ранее, диссоциация цитохрома c D19, образованного непосредственно из раствора, приводит к узкому симметричному процессу разделения заряда [56]. МС / МС D17, образованного непосредственно из раствора, приводит к относительно широкому, но симметричному процессу разделения заряда (преимущественно D17 → M9 + M8) ().
МС / МС спектры ( a ) (димеры цитохрома c + 17H) 17+ , (D17), образованные непосредственно ESI, ( b ) (димеры цитохрома c + 13H) 13+ , (D13), образованный выделением D19-D17, восстановлением газофазного переноса протона диэтиламином, выделением и последующей активацией D13, и ионы ( c ) D13, образованные непосредственно из раствора.Ионы образовывались из 100 мМ раствора (вода: метанол, 1: 1 + 2% уксусная кислота).
Напротив, диссоциация D13, образованного непосредственно из раствора, приводит к сильно асимметричному распределению заряда с центром вокруг D13 → M9 + M4 (). Ионы D17-D19 были изолированы и прореагировали с триэтиламином внутри ионной ячейки FT-ICR в течение 100 с, что привело к образованию преимущественно ионов D13. Выделение D13 и последующая активация этих ионов дает спектр МС / МС, показанный на. Доминирующим процессом является симметричная диссоциация с центром D13 → M7 + M6.Таким образом, процесс диссоциации D13 явно зависит от того, образуются ли они в результате восстановления с переносом протона в газовой фазе D17 – D19 () или образуются непосредственно из раствора (). В этих экспериментальных условиях внутренняя энергия иона уравновешивается с полем излучения черного тела при комнатной температуре до активации иона [63]. Таким образом, различие во фрагментации не может быть артефактом различия внутренней энергии. Эти результаты показывают, что структуры ионов D13 различаются для этих двух методов образования ионов и что структура димера влияет на то, как эти димеры диссоциируют, т.е.е., через симметричное и асимметричное разделение заряда. Следует отметить, что димеризация в растворе неспецифична и что может присутствовать большой ансамбль димерных структур. Однако ясно, что существуют по крайней мере две отдельные структуры или различные ансамбли структур.
«Память» ионов D13 может быть связана с различными димерными структурами, существующими в растворе. Эти димеры могут получать разное количество зарядов в процессе ионизации электрораспылением, или процесс электрораспыления может вызывать конформационные изменения в некоторой части димеров при переходе из раствора в газовую фазу, что приводит к различным структурам димеров для D17 по сравнению с D13.Мы не можем четко различать эти две возможности, основываясь только на этих результатах. Однако широкое распределение в зарядовых состояниях димеров, образованных в этих денатурирующих условиях, по сравнению с типичным единственным доминирующим зарядовым состоянием димера, наблюдаемым при электрораспылении из растворов, в которых эти молекулы имеют нативную структуру, предполагает, что первый механизм более вероятен, т. Е. в растворе существуют по крайней мере две отдельные структуры или ансамбль структур, и эти структуры в конечном итоге имеют разную степень заряда в процессе электрораспыления.
Аналогичные результаты наблюдаются для ионов D15, образованных из раствора, по сравнению с ионами, образованными переносом протона. Те, которые образуются непосредственно из раствора, демонстрируют бимодальную диссоциацию: симметричный процесс, сосредоточенный вокруг M8 / M7, и асимметричный процесс, сосредоточенный вокруг M10 / M5 (). Восстановление заряда D19 – D17 с образованием более низких зарядовых состояний с последующим выделением и диссоциацией D15 приводит только к симметричной диссоциации с образованием M8 / M7 (). Опять же, эти ионы D15 явно сохраняют «память» о том, образовались ли они в газовой фазе из более высоких зарядовых состояний, которые симметрично диссоциируют, или непосредственно из раствора.
МС / МС спектры (цитохрома c димеры + 15H) 15+ , (D15), образованного ( a ) непосредственно ESI и ( b ), образованного выделением D19-D17, газ- восстановление фазового переноса протона диэтиламином, выделение и последующая активация D15. Ионы образовывались из 100 мМ раствора (вода: метанол, 1: 1 + 2% уксусная кислота).
Газофазный H / D обмен мономеров и димеров цитохрома c с ND
3Предыдущие исследования показали, что степень асимметрии заряда цитохрома c D11, образованного непосредственно наноэлектрораспылением с использованием водных растворов, содержащих 100 мМ ацетата аммония нейтральный pH зависел от внутренней энергии, которая вкладывалась в ионы [56].Чтобы попытаться установить, в какой степени различные конформеры могут вносить вклад в наблюдаемую энергетическую зависимость, на D11 были проведены исследования обмена H / D. Тщательная настройка параметров источника ESI, в частности увеличение напряжения смещения на внешнем гексаполе накопления и увеличение времени нагрузки гексаполя, способствует образованию цитохрома c D11 ().
ESI масс-спектр 500 мкМ M конский цитохром c в водном растворе со 100 мМ ацетатом аммония, pH ~ 7, со спектрометром, настроенным на получение максимального количества димеров с зарядами 11+ (D11).
Для этих экспериментов по газофазному H / D обмену давление NH 3 изменялось от 1 × 10 –8 до 3 × 10 –7 торр с постоянным временем реакции 180 с. Для эксперимента при самом высоком давлении реагента (наибольшая степень обмена H / D) () среднее количество атомов водорода, обмениваемых на дейтерий, составляет ~ 170 после вычитания обмена 11 протонов. Ширина распределения составляет примерно 110 Да. Отношение сигнал / шум недостаточно, чтобы приписать вариации интенсивностей изотопов разрешимым конформерам.Напротив, M7 в тех же условиях показывает средний обмен ~ 84 атомов водорода на дейтерий с шириной изотопического распределения ~ 50. Таким образом, как средняя степень обмена, так и ширина распределения димера примерно вдвое больше, чем у мономера. Если бы присутствовала только одна структура, нельзя было бы ожидать, что суммарная максимальная степень обмена и ширина распределения будут линейно увеличиваться с массой иона, если предположить, что степень обмена H / D связана с открытой поверхностью кластера.Эти результаты предполагают присутствие множественных конформеров димеров, которые не разрешаются в этих экспериментах по обмену H / D, что согласуется с нашим ожиданием, что эти ионы D11 образуются в результате неспецифической димеризации молекул циктохрома c в растворе.
Частичные масс-спектры ESI цитохрома c (показана область D11) при тех же условиях раствора, что и раньше, и после газофазного водородно-дейтериевого обмена с ND 3 в течение 180 с при ( b ) 1 × 10 −7 торр и ( c ) 3 × 10 −7 торр.
Обширные исследования газофазного H / D обмена мономеров цитохрома c были проведены McLafferty и соавторами [22–24], которые обнаружили целых семь конформаций цитохрома c M7, образованных непосредственно ESI [24]. В наших экспериментах в качестве обменного реагента используется ND 3 вместо D 2 O [24], и время реакции короче. Распределение изотопов для M7 предполагает наличие нескольких конформеров, согласующихся с результатами, полученными Маклафферти и соавторами.
Выводы
Асимметричное распределение заряда, которое наблюдается при диссоциации гомодимеров белков, можно объяснить двумя различными механизмами. В одном механизме асимметрия индуцируется разворачиванием одного из белков в димере при активации. В димерах, состоящих из двух почти идентичных белков, белок с более высокой конформационной гибкостью отделяется от комплекса с большим зарядом. Это может быть связано с тем, что часть молекулы покидает барьер диссоциации, где дальнейшее развертывание молекулы индуцируется отталкивающим кулоновским потенциалом.Во время этого процесса разворачивания и разделения протоны передаются разворачивающемуся белку, чтобы уменьшить общую кулоновскую энергию. Этот механизм лучше всего объясняет результаты, наблюдаемые для гетеродимеров α -лактабумина, состоящих из одного окисленного и одного восстановленного белка. Этот механизм также лучше всего объясняет результаты для димеров цитохрома c с 11 зарядами, для которых наблюдается энергетическая зависимость в степени асимметрии [56].
Некоторая часть наблюдаемого асимметричного распределения заряда также может быть приписана другому механизму, в котором структуры мономеров в димере различаются.Это лучше всего объясняет результаты, наблюдаемые для димеров цитохрома c с зарядами 17+ по сравнению с димерами с 13+ зарядами. При образовании непосредственно из раствора первый диссоциирует с симметричным распределением заряда, тогда как последний диссоциирует с сильно асимметричным распределением заряда. Однако, когда зарядовое состояние ионов димера 17+ снижается до 13+ в газовой фазе посредством мягких реакций переноса протона, образующиеся ионы 13+ диссоциируют с симметричным распределением заряда.Это ясно демонстрирует, что ионы 13+, образованные из ионов 17+, имеют другую структуру (или разные ансамбли структур), чем те, которые образуются непосредственно из раствора, и что это структурное различие отвечает за асимметричное и симметричное распределение заряда, наблюдаемое при диссоциации этих 13+. ионы. Это первое зарегистрированное свидетельство того, что асимметричное распределение заряда может быть вызвано структурными различиями в субъединицах мономера в комплексе.
Наконец, H / D обмен зарядового состояния 11+ димеров цитохрома c указывает на присутствие множественных конформеров, хотя эти конформеры не разрешаются в этом эксперименте.Дальнейшая работа с другими реагентами для обмена H / D или в других условиях, или с конформационно-селективными методами, такими как подвижность ионов, в сочетании с экспериментами по диссоциации может предоставить дополнительную полезную информацию о процессе разделения заряда. Лучшее понимание этого процесса в конечном итоге улучшит структурную информацию, которая может быть получена при газофазной диссоциации нековалентных комплексов.
Благодарности
Авторы благодарят доктора Дэвида Кинга (Калифорнийский университет, Беркли) за восстановление и очистку α -лактальбумина.Эта работа была поддержана Национальным институтом здравоохранения (грант № R01-GM64712-01).
Ссылки
1. Калета Д.Т., Джарролд М.Ф. Пептидные вертушки. J Am Chem Soc. 2002; 124: 1154–1155. [PubMed] [Google Scholar] 2. Kaleta DT, Jarrold MF. Нековалентные взаимодействия между несольватированными пептидами. J. Phys Chem A. 2002; 106: 9655–9664. [Google Scholar] 3. Clemmer DE, Jarrold MF. Измерения подвижности ионов и их приложения к кластерам и биомолекулам. J. Mass Spectrom. 1997. 32: 577–592. [Google Scholar] 4.Counterman AE, Valentine SJ, Srebalus CA, Henderson SC, Hoaglund CS, Clemmer DE. Структура высокого порядка и диссоциация газообразных пептидных агрегатов, скрытых в масс-спектрах. J Am Soc масс-спектрометрия. 1998. 9: 743–759. [PubMed] [Google Scholar] 5. Counterman AE, Hilderbrand AE, Barnes CAS, Clemmer DE. Формирование пептидных агрегатов во время ESI: размер, заряд, состав и вклад в шум. J Am Soc масс-спектрометрия. 2001; 12: 1020–1035. [Google Scholar] 6. Гидден Дж., Виттенбах Т., Батька Дж. Дж., Вайс П., Джексон А. Т., Скривенс Дж. Х., Бауэрс МТ.Энергетика сворачивания и динамика макромолекул в газовой фазе: щелочные ионно-катионизированные олигомеры поли (этилентерефталата). J Am Chem Soc. 1999; 121: 1421–1422. [Google Scholar] 7. Гевремонт Р., Purves RW. Спектрометрия-масс-спектрометрия подвижности ионов с асимметричной формой волны в высоком поле: исследование ионов лейцина-энкефалина, полученных с помощью ионизации электрораспылением. J Am Soc масс-спектрометрия. 1999; 10: 492–501. [PubMed] [Google Scholar] 8. Purves RW, Barnett DA, Guevremont R. Разделение белков-конформеров с использованием спектрометрии-масс-спектрометрии асимметричной формы волны с асимметричной формой волны электроспрея.Int J масс-спектрометрия. 2000; 197: 163–177. [Google Scholar] 9. Дэн Ю.З., Чжан З.К., Смит Д.Л. Сравнение непрерывного и импульсного мечения амидного водородного обмена / масс-спектрометрии для исследования динамики белков. J Am Soc масс-спектрометрия. 1999; 10: 675–684. [PubMed] [Google Scholar] 10. Чжу М.М., Ремпель Д.Л., Брутто М.Л. Данные моделирования титрования, амидного H / D обмена и масс-спектрометрии для получения констант связывания белок-лиганд. J Am Soc масс-спектрометрия. 2004. 15: 388–397. [PubMed] [Google Scholar] 11. Гарда Э., Уилларда Д., Брегара Дж., Грин МК, Лебрилла CB.Сайт-специфичность в H-D-обменных реакциях газофазных протонированных аминокислот с Ch4OD. Org Mass Spectrom. 1993; 28: 1632–1639. [Google Scholar] 12. Кэмпбелл С., Роджерс М. Т., Марцлафф Е. М., Бошам Дж. Л.. Структурные и энергетические ограничения на реакции газофазного водородно-дейтериевого обмена протонированных пептидов с D2O, CD3OD, CD3CO2D и ND3. J Am Chem Soc. 1994; 116: 9765–9766. [Google Scholar] 13. Gard E, Green MK, Bregar J, Lebrilla CB. Газофазный водород-дейтериевый обмен как молекулярный зонд для взаимодействия метанола и протонированных пептидов.J Am Soc масс-спектрометрия. 1994; 5: 623–631. [PubMed] [Google Scholar] 14. Кэмпбелл С., Роджерс М. Т., Марцлафф Е. М., Бошам Дж. Л.. Реакции обмена дейтерия как зонд структуры биомолекул. Фундаментальные исследования газофазных реакций H / D обмена протонированных олигомеров глицина с D2O, CD3OD, CD3CO2D и ND3. J Am Chem Soc. 1995; 117: 12840–12854. [Google Scholar] 15. Грин МК, Penn SG, Lebrilla CB. Комплексообразование протонированных пептидов с сахаридами в газовой фазе снижает скорость реакций обмена водород / дейтерий.J Am Soc масс-спектрометрия. 1995; 6: 1247–1251. [PubMed] [Google Scholar] 16. Калташов И.А., Дорошенко В.М., Коттер Р.Дж. Газофазные реакции обмена пептидных ионов водород / дейтерий в квадрупольном масс-спектрометре с ионной ловушкой. Белки. 1997. 28: 53–58. [PubMed] [Google Scholar] 17. Heck AJR, Jorgensen TJD, O’Sullivan M, von Raumer M, Derrick PJ. Газофазные нековалентные взаимодействия между антибиотиками группы ванкомицина и пептидами-предшественниками бактериальной клеточной стенки, вызванные обменом водорода / дейтерия. J Am Soc масс-спектрометрия.1998. 9: 1255–1266. [Google Scholar] 18. Фрейтас М.А., Маршалл АГ. Скорость и степень газофазного водородно-дейтериевого обмена брадикининов: данные о пептидных цвиттерионах в газовой фазе. Int J масс-спектрометрия. 1999. 183: 221–231. [Google Scholar] 19. Рейзер ML, Brodbelt JS. Газофазные реакции обмена H / D полиаминовых комплексов: [M + H) (+), (M плюс щелочной металл (+)] и (M + 2H) (2+) J Am Soc Mass Spectrom. 2000; 11 : 711–721. [PubMed] [Google Scholar] 20. Леви-Сери Э, Костер Г., Коган А., Гутман К., Рубен Б. Г., Лифшиц К.Исследование H / D-обмена в протонированном брадикинине с помощью ионизационной трубки с электрораспылением. J. Phys Chem A. 2001; 105: 5552–5559. [Google Scholar] 21. Солоуки Т., Форт Р.С., Аломари А., Фаттахи А. Реакции газофазного водородно-дейтериевого обмена модельного пептида: FT-ICR и вычислительный анализ конформационных мутаций, индуцированных металлами. J Am Soc масс-спектрометрия. 2001; 12: 1272–1285. [PubMed] [Google Scholar] 22. Suckau D, Shi Y, Beu SC, Senko MW, Quinn JP, Wampler FM, McLafferty FW. Сосуществующие стабильные конформации газообразных белковых ионов.Proc Nat Acad Sci USA. 1993; 90: 790–793. [Бесплатная статья PMC] [PubMed] [Google Scholar] 23. Wood TD, Chorush RA, Wampler FM, Little DP, O’Connor PB, McLafferty FW. Газофазное сворачивание и разворачивание катионов цитохрома- c . Proc Natl Acad Sci USA. 1995; 92: 2451–2454. [Бесплатная статья PMC] [PubMed] [Google Scholar] 24. McLafferty FW, Guan ZQ, Haupts U, Wood TD, Kelleher NL. Газообразные конформационные структуры цитохрома c. J Am Chem Soc. 1998. 120: 4732–4740. [Google Scholar] 25. Рид Г.Е., О’Хэр RAJ, Styles ML, McFadyen WD, Simpson RJ.Газофазные ионно-молекулярные реакции в модифицированной ионной ловушке: H / D-обмен нековалентных комплексов и координационно-ненасыщенных комплексов платины. Масс-спектрометр Rapid Commun. 1998; 12: 1701–1708. [Google Scholar] 26. Коган А., Устюжанин П., Рубен Б. Г., Лифшиц С. Водород / дейтериевый обмен мономеров и димеров лейцин-энкефалина. Int J масс-спектрометрия. 2002; 213: 1–4. [Google Scholar] 27. Мао Д., Дуглас ди-джей. H / D-обмен газофазными ионами брадикинина в линейной квадрупольной ионной ловушке. J Am Soc масс-спектрометрия.2003. 14: 85–94. [PubMed] [Google Scholar] 28. Ли SW, Ли Х.Н., Ким Х.С., Бошам Дж. Л.. Селективное связывание краун-эфиров с протонированными пептидами может быть использовано для исследования механизмов обмена H / D и реакций диссоциации, вызванных столкновениями, в газовой фазе. J Am Chem Soc. 1998. 120: 5800–5805. [Google Scholar] 29. Валовой DS, Schnier PD, Rodriguezcruz SE, Fagerquist CK, Williams ER. Конформации и сворачивание ионов лизоцима в вакууме. Proc Nat Acad Sci USA. 1996; 93: 3143–3148. [Бесплатная статья PMC] [PubMed] [Google Scholar] 31.Loo JA. Изучение нековалентных белковых комплексов методом масс-спектрометрии с ионизацией электрораспылением. Масс-спектром. Ред. 1997; 16: 1–23. [PubMed] [Google Scholar] 32. Лим Х.К., Сие Й.Л., Ганем Б., Хенион Дж. Распознавание пептидных лигандов клеточной стенки антибиотиками группы ванкомицина – исследования с использованием масс-спектрометрии с ионным распылением. J. Mass Spectrom. 1995; 30: 708–714. [Google Scholar] 33. Лоо Дж. А., Ху П. Ф., МакКоннелл П., Мюллер В. Т., Сойер Т. К., Танабал В. Исследование взаимодействий связывания белка с фосфопептидом домена Src Sh3 с помощью масс-спектрометрии с ионизацией электрораспылением.J Am Soc масс-спектрометрия. 1997. 8: 234–243. [Google Scholar] 34. Йоргенсен TJD, Roepstorff P, Heck AJR. Прямое определение констант связывания раствора для нековалентных комплексов между пептидными аналогами бактериальной клеточной стенки и антибиотиками группы ванкомицина методом масс-спектрометрии с ионизацией электрораспылением. Anal Chem. 1998; 70: 4427–4432. [Google Scholar] 35. Дэниел Дж. М., Фрисс С. Д., Раджагопалан С., Вендт С., Зеноби Р. Количественное определение нековалентных связывающих взаимодействий с использованием масс-спектрометрии с мягкой ионизацией.Int J масс-спектрометрия. 2002; 216: 1-27. [Google Scholar] 36. Валовой DS, Чжао YX, Уильямс ER. Диссоциация комплексов гем-глобин с помощью инфракрасной радиационной диссоциации черного тела: молекулярная специфичность в газовой фазе? J Am Soc масс-спектрометрия. 1997. 8: 519–524. [Бесплатная статья PMC] [PubMed] [Google Scholar] 37. Schnier PD, Klassen JS, Strittmatter EE, Williams ER. Энергии активации для диссоциации двухцепочечных олигонуклеотидных анионов: данные о спаривании оснований Уотсона-Крика в вакууме. J Am Chem Soc. 1998; 120: 9605–9613.[Бесплатная статья PMC] [PubMed] [Google Scholar] 38. Рогалевич Ф., Хоппиллиард Я., Оханесян Г. Структуры и фрагментация комплексов цинка (II) аминокислот в газовой фазе. IV. Влияние растворителя на структуру электрораспыленных ионов. Int J масс-спектрометрия. 2003. 227: 439–451. [Google Scholar] 39. Габелика В., Де Пау Э. Сравнение между фазовой стабильностью в растворе и кинетической стабильностью в газовой фазе олигодезоксинуклеотидных дуплексов. J. Mass Spectrom. 2001; 36: 397–402. [PubMed] [Google Scholar] 40. Габелика V, Де Пау Э.Сравнение индуцированной столкновением диссоциации дуплексной ДНК при различных режимах коллизии: доказательства многоступенчатого механизма диссоциации. J Am Soc масс-спектрометрия. 2002; 13: 91–98. [PubMed] [Google Scholar] 41. Габелика В., Де Пау Э. Диссоциация 16-мерных дуплексов ДНК с различными последовательностями, вызванная столкновением: свидетельства сохранения конформации двойной спирали в газовой фазе. Int J масс-спектрометрия. 2002; 219: 151–159. [Google Scholar] 42. Ван К.Х., Шибуэ Т., Брутто ML. Нековалентные комплексы между ДНК-связывающими лекарствами и двухцепочечными олигодезоксинуклеотидами: исследование методом масс-спектрометрии с ионной ловушкой ESI.J Am Chem Soc. 2000; 122: 300–307. [Google Scholar] 43. Чен Ю.Л., Кэмпбелл Дж. М., Коллингс Б. А., Конерманн Л., Дуглас Д. Д.. Устойчивость высокозаряженного нековалентного комплекса в газовой фазе: голомиоглобина. Масс-спектрометр Rapid Commun. 1998. 12: 1003–1010. [PubMed] [Google Scholar] 44. Хантер С.Л., Маук А.Г., Дуглас Диджей. Диссоциация гема от миоглобина и цитохрома b (5): сравнение поведения в растворе и газовой фазе. Биохим США. 1997; 36: 1018–1025. [PubMed] [Google Scholar] 45. Йоргенсен Т.Д., Делфорж Д., Ремакл Дж., Бойсен Дж., Рёпсторф П.Индуцированная столкновением диссоциация нековалентных комплексов между антибиотиками ванкомицина и стереоизомерами пептидного лиганда: доказательства молекулярного распознавания в газовой фазе. Int J масс-спектрометрия. 1999. 188: 63–85. [Google Scholar] 46. Loo JA, He JX, Cody WL. Структура высшего порядка в газовой фазе отражает структуру раствора. J Am Chem Soc. 1998; 120: 4542–4543. [Google Scholar] 47. Ростом А.А., Фучини П., Бенджамин Д.Р., Юенеманн Р., Нирхаус К.Х., Хартл Ф.У., Добсон С.М., Робинсон К.В. Обнаружение и селективная диссоциация интактных рибосом в масс-спектрометре.Proc Nat Acad Sci USA. 2000; 97: 5185–5190. [Бесплатная статья PMC] [PubMed] [Google Scholar] 48. Pinkse MWH, Maier CS, Kim JI, Oh BH, Heck AJR. Макромолекулярная оценка уреазы Helicobacter pylori, исследованная методом масс-спектрометрии. J. Mass Spectrom. 2003. 38: 315–320. [PubMed] [Google Scholar] 49. Лайт-Вал К.Дж., Шварц Б.Л., Смит Р.Д. Наблюдение нековалентных четвертичных ассоциаций белков с помощью масс-спектрометрии с ионизацией электрораспылением. J Am Chem Soc. 1994; 116: 5271–5278. [Google Scholar] 50. Шварц Б.Л., Брюс Дж. Э., Андерсон Г. А., Хофстадлер С. А., Роквуд А. Л., Смит Р. Д., Чилкоти А., Стейтон П. С..Диссоциация тетрамерных ионов нековалентных стрептавидиновых комплексов, образующихся при ионизации электрораспылением. J Am Soc масс-спектрометрия. 1995; 6: 459–465. [PubMed] [Google Scholar] 51. Фицджеральд М.С., Чернушевич И., Стэндинг К.Г., Уитмен С.П., Кент SBH. Исследование олигомерной структуры фермента методом времяпролетной масс-спектрометрии с ионизацией электрораспылением. Proc Nat Acad Sci USA. 1996; 93: 6851–6856. [Бесплатная статья PMC] [PubMed] [Google Scholar] 52. Фелицын Н., Китова Е.Н., Классен Я.С. Термическое разложение газообразного мультибелкового комплекса, исследованное методом черной инфракрасной радиационной диссоциации.Исследование происхождения асимметричного поведения диссоциации. Anal Chem. 2001; 73: 4647–4661. [PubMed] [Google Scholar] 53. Бенеш JLP, Соботт Ф., Робинсон CV. Термическая диссоциация мультимерных белковых комплексов с помощью масс-спектрометрии с наноэлектрораспылением. Anal Chem. 2003. 75: 2208–2214. [PubMed] [Google Scholar] 55. Versluis C, van der Staaij A, Stokvis E, Heck AJR, de Craene B. Образование метастабильных ионов и разделение разрозненных зарядов при газофазной диссекции белковых сборок, изученных методом ортогональной времяпролетной масс-спектрометрии.J Am Soc масс-спектрометрия. 2001; 12: 329–336. [PubMed] [Google Scholar] 56. Jurchen JC, Williams ER. Происхождение асимметричного разделения заряда при диссоциации газофазных белковых гомодимеров. J Am Chem Soc. 2003; 125: 2817–2826. [Бесплатная статья PMC] [PubMed] [Google Scholar] 57. Jurchen JC, Garcia DE, Williams ER. Пути газофазной диссоциации многозарядных пептидных кластеров. J Am Soc масс-спектрометрия. 2003. 14: 1373–1386. [Бесплатная статья PMC] [PubMed] [Google Scholar] 58. Готье Дж. В., Траутман Т. Р., Якобсон Д.Б.Продолжительное нерезонансное облучение для диссоциации, активируемой столкновениями, с использованием метода масс-спектрометрии с преобразованием Фурье и диссоциации, активируемой столкновениями, которая имитирует инфракрасную многофотонную диссоциацию. Анальный Чим Акта. 1991; 246: 211–225. [Google Scholar] 59. Вингер Б.Э., Лайт-Валь КДЖ, Роквуд А.Л., Смит Р.Д. Исследование качественных различий конформации многократно протонированных газофазных белков с помощью изотопного обмена H / D с D2O. J Am Chem Soc. 1992; 114: 5897–5898. [Google Scholar] 60. Фелицын Н., Китова Е.Н., Классен Я.С.Термическая диссоциация гомодимера белка экотина в газовой фазе. J Am Soc масс-спектрометрия. 2002; 13: 1432–1442. [PubMed] [Google Scholar] 61. Апостол I. Оценка относительной стабильности сконструированных гемоглобинов с помощью масс-спектрометрии с электрораспылением. Анальная биохимия. 1999; 272: 8–18. [PubMed] [Google Scholar] 62. Versluis C, Heck AJR. Газофазная диссоциация гемоглобина. Int J масс-спектрометрия. 2001; 210: 637–649. [Google Scholar] 63. Цена WD, Williams ER. Активация пептидных ионов излучением черного тела: факторы, приводящие к кинетике диссоциации в пределе быстрого обмена энергией.J. Phys Chem A. 1997; 101: 8844–8852. [Бесплатная статья PMC] [PubMed] [Google Scholar]Дальнейшие исследования происхождения асимметричного разделения заряда в белковых гомодимерах
J Am Soc Mass Spectrom. Авторская рукопись; доступно в PMC 2006 6 января.
Опубликован в окончательной редакции как:
PMCID: PMC1325214
NIHMSID: NIHMS3986
Химический факультет Калифорнийского университета в Беркли, Беркли, Калифорния, США
Запросы на перепечатку адресов: ДокторЭ. Р. Уильямс, химический факультет Калифорнийского университета в Беркли, Беркли, Калифорния 94720, США. Электронное письмо: ude.yelekreb.mehcc@smailliw См. другие статьи в PMC, в которых цитируется опубликованная статья.Abstract
Диссоциация протонированных в газовой фазе белковых димеров на составляющие их мономеры может приводить к симметричному или асимметричному распределению заряда. Диссоциация гомодимеров α -лактальбумина с 15+ зарядами приводит к симметричному, но широкому распределению белковых мономеров с зарядовыми состояниями, сосредоточенными вокруг 8 + / 7 +.Напротив, диссоциация гетеродимера 15+, состоящего из одной молекулы в окисленной форме и одной в восстановленной форме, приводит к сильно асимметричному распределению зарядов, при котором восстановленные частицы уносят преимущественно заряды 11+, а окисленная молекула уносит заряды 4+. . Этот результат не может быть адекватно объяснен дифференциальной зарядкой, происходящей ни в растворе, ни в процессе электрораспыления, но, по-видимому, лучше всего объясняется восстановленными частицами, разворачивающимися при активации в газовой фазе с последующим разделением и переносом протонов на разворачивающиеся частицы в диссоциативном комплексе. чтобы минимизировать кулоновское отталкивание.Для димеров цитохрома c , образованных непосредственно из раствора, зарядовое состояние 17+ подвергается симметричному распределению заряда, тогда как диссоциация 13+ асимметрична. Уменьшение зарядового состояния димеров с зарядами 17+ до 13+ посредством переноса протона в газовой фазе и последующей диссоциации выбранных по массе ионов 13+ приводит к симметричному распределению заряда. Этот результат ясно показывает, что структура димерных ионов с зарядами 13+ зависит от метода образования иона и что структурные различия ответственны за наблюдаемое симметричное и асимметричное разделение зарядов.Это указывает на то, что асимметрия, наблюдаемая, когда эти ионы образуются непосредственно из раствора, должна происходить либо из-за различий в конформациях мономера в димере, которые существуют в растворе, либо в процессе ионизации электрораспылением. Эти результаты предоставляют дополнительные доказательства происхождения асимметрии заряда, которая возникает при диссоциации многозарядных белковых комплексов, и показывают, что некоторая информация о фазе раствора может быть получена из этих экспериментов по газофазной диссоциации.
Биологическая функциональность белков и белковых комплексов сильно зависит от их структур в фазе раствора. Важность области структурной биологии отражена в нескольких недавно присужденных Нобелевских премиях за вклад в методы структурного выяснения, включая ЯМР (Курт Вутрих, 2002 г. по химии), рентгеновскую кристаллографию (Иоганн Дайзенхофер, Роберт Хубер и Хартмут Мишель, 1988 г. в. химия; Герберт Хауптман и Джером Карл 1985 по химии) и кристаллографическая электронная микроскопия (сэр Аарон Клаг 1982 по химии).Анализ белков и белковых комплексов этими методами может предоставить подробную трехмерную информацию, но эти методы часто ограничены экспериментальными ограничениями, такими как необходимость получения полезных кристаллов белка или требование значительного количества образца. Напротив, для масс-спектрометрии требуется небольшой образец, а с ионизацией электрораспылением (ESI) или мягкой лазерной десорбцией / ионизацией (John Fenn and Kiochi Tanaka, 2002 по химии) в масс-спектрометр можно ввести большое количество биомолекул и специфических комплексов.Был разработан ряд методов для исследования общих трехмерных форм ионов биомолекул и нековалентных комплексов с использованием методов ионной подвижности [1–8], H / D обмена как в растворе [9, 10], так и в газовой фазе [ 11–28] и реакционной способности с переносом протона [29, 30].
Для анализа нековалентных комплексов масс-спектрометрия имеет то преимущество, что стехиометрию комплекса можно определить с помощью простого измерения массы [31]. В некоторых случаях MS может обеспечить правильные константы связывания в фазе раствора для белок-лигандных взаимодействий [32–35].Эти разработки подробно рассмотрены [31, 35]. В принципе, информацию о структуре нековалентных комплексов можно потенциально получить, исследуя ионы продуктов, образующиеся в результате диссоциации комплекса, в эксперименте тандемной масс-спектрометрии (МС / МС). Объем соответствующей информации, которую можно получить в результате этих экспериментов, зависит от количества структурной информации о фазе раствора, которая сохраняется биологическими комплексами при переходе в газовую фазу (или от того, может ли эта информация быть восстановлена на основе сведений о происходящих структурных изменениях. во время этого перехода) и степень, в которой методы MS / MS нарушают любую оставшуюся структурную информацию.Многие эксперименты показывают, что газофазные комплексы биомолекул и даже некоторые небольшие молекулы могут сохранять «память» о своей фазовой структуре раствора, и что эта память может быть исследована процессами диссоциации [36–48].
Ряд исследований гомогенных биологических комплексов (тетрамеры, пентамеры, гексамеры и т. Д.) С помощью МС / МС выявили первоначально неожиданный результат, заключающийся в том, что первичный путь диссоциации мультимерных комплексов представляет собой выброс высоко заряженного мономера, который удаляет 30-50 % заряда комплекса [49–53].Рентгеновская кристаллография показывает, что структуры растворов субъединиц во многих гомогенных комплексах идентичны. В некоторых случаях весь комплекс кристаллизуется внутри асимметричной единицы кристалла, как в случае с шига-подобным токсином [9], в то время как в других, таких как апострептавидин, асимметричная единица содержит одну субъединицу, и структура комплекс экстраполирован с помощью молекулярного моделирования [54]. Из-за практически идентичных структур фазы раствора участвующих субъединиц в этих биологических комплексах наблюдаемое асимметричное распределение заряда, по-видимому, является артефактом процедуры ESI-MS / MS.Было высказано предположение, что асимметричное распределение заряда коррелирует с газофазными конформационными перестройками [50, 52, 55, 56]. Также было продемонстрировано, что степень асимметричного распределения заряда для гомодимеров белков зависит от конформационной гибкости белков в комплексе, зарядового состояния комплекса, отложения внутренней энергии и состава растворов для электрораспыления, из которых комплексы образуются. формируются [56]. Для более крупных кластеров, состоящих из идентичных субъединиц, наблюдалось деление с преимущественным образованием протонированных димеров, а с увеличением зарядового состояния и размера кластера наблюдалось усиление образования протонированного мономера [57].Степень деления по сравнению с потерями нейтралов для этих кластеров может быть связана с наблюдаемой для многозарядных металлических кластеров с одним важным отличием: различия в конформационной гибкости субъединиц мономера внутри кластера приводят к различиям в коэффициентах ветвления различных диссоциаций. процессы, наблюдаемые для этих многократно протонированных кластеров биомолекул [57].
Газофазный водород-дейтериевый (H / D) обмен обычно осуществляется путем хранения интересующего иона в вакууме и измерения изменения массы иона из-за реакций обмена H / D, которые происходят во время столкновений с нейтральными молекулами обменного реагента. , например, D 2 O или ND 3 .Хотя газофазный обмен H / D широко использовался для характеристики пептидов и малых молекул [11–21], а также структуры некоторых газофазных белков [22–24], очень мало исследований было выполнено с нековалентными комплексами, включающими биологические ионы, и эта нековалентная работа была сосредоточена на димерах пептидов [25, 26] и пептидных комплексах с ионами металлов [16, 18, 21, 27] и небольшими молекулами [15, 28].
Здесь представлена дальнейшая характеристика гомодимеров белка с использованием газофазной диссоциации и газофазного H / D обмена.Исследования диссоциации смешанных димеров окисленного и восстановленного α -лактальбумина предоставляют дополнительные доказательства того, что конформационная гибкость играет ключевую роль в определении степени асимметричного распределения заряда. Для диссоциации гомодимеров цитохрома c показано, что зарядовые состояния, полученные в результате снижения заряда в газовой фазе из гомодимеров с более высоким зарядовым состоянием, диссоциируют совсем иначе, чем состояния, полученные непосредственно из раствора. Представлена первая демонстрация газофазного H / D обмена белкового комплекса.Эти результаты показывают, что два разных механизма ответственны за асимметричное распределение заряда, наблюдаемое при диссоциации гомодимеров белка.
Experimental
Эксперименты проводились на масс-спектрометре Berkeley-Bruker с узким проходом 9,4 Тл с ионно-циклотронным резонансом с преобразованием Фурье (FT-ICR), который описан в другом месте [56]. Используемые экспериментальные процедуры аналогичны использованным ранее [56]. Краткое описание и дополнительные сведения приведены ниже.
Chemicals
Цитохром сердца лошади c , α -лактальбумин из коровьего молока и триэтиламин были приобретены у Sigma-Aldrich Co. (Сент-Луис, Миссури) и использовались без дополнительной очистки. Дейтерированный аммиак был приобретен в Cambridge Isotope Laboratories (Woburn, MA). Ацетамидирование α -лактальбумина было выполнено доктором Дэвидом Кингом (Калифорнийский университет, Беркли). Все четыре внутримолекулярные дисульфидные связи α -лактальбумина восстанавливаются трис (карбоксиэтил) фосфином, ацетамидируются в темноте йодацетамидом в течение одного часа и очищаются с помощью ВЭЖХ для удаления буфера и непрореагировавших реагентов.Практически полное восстановление и ацетамидирование всех дисульфидных связей в α -лактальбумине было подтверждено измерениями массы.
Образование димеров
Образование неспецифических белковых димеров усиливается за счет использования концентраций белка 100–500 мкМ M и регулирования напряжений на границе раздела для «мягкого» введения ионов. Сигнал димера улучшается за счет настройки на селективное накопление ионов во внешнем гексаполе в области источника прибора. Это достигается накоплением ионов в гексаполе в течение 3–4 с (против ≤ 1.0 с), увеличивая смещение постоянного тока гексаполя с ~ 2,7 В до 3,5–4,5 В и вводя несколько скоплений гексаполя в ионную ячейку перед обнаружением.
Смешанные димеры окисленного (все четыре дисульфидные связи не повреждены) и восстановленного (все четыре дисульфидные связи разорваны и все восемь остатков цистеина впоследствии ацетамидированы) α -лактальбумин были образованы путем смешивания растворов двух форм α -лактальбумина в различных соотношения. Концентрация общего белка поддерживалась постоянной на уровне ~ 200 мкМ М, и отношения двух компонентов были изменены, чтобы максимизировать абсолютное содержание смешанного димера.Наибольшее количество смешанного димера появляется при относительной концентрации восстановленного до окисленного α -лактальбумина ~ 2: 1. Поскольку концентрации белка определялись массой лиофилизированных порошков, используемых для приготовления раствора, эти концентрации являются приблизительными. Ионы димера белка формировали с использованием ESI со скоростью потока ~ 1 мкМ л / мин в условиях раствора 1: 1 вода: метанол + 2% уксусная кислота.
Димеры цитохрома c были сформированы с использованием обычного ESI при скорости потока 1-2 мкМ л / мин и концентрации 100 мкМ М в 1: 1 вода: метанол + 2% уксусная кислота.Зарядовые состояния димеров, сформированные непосредственно из раствора, варьируются от [(цитохром c ) 2 + 19H] 19+ до [(циктохром c ) 2 + 13H] 13+ (сокращенно D19 – D13) . Зарядовые состояния димера с m / z выше, чем у D17, были выброшены из ионной ячейки с использованием коррелированных выстрелов, оставляя зарядовые состояния D19 – D17, оставшиеся в ионной ячейке. Вакуумная камера FT-ICR была подготовлена для снижения заряда путем кратковременного введения триэтиламина в вакуумную камеру под давлением ~ 3 × 10 90 · 10 7 -8 90 · 108 торр с использованием пьезоэлектрического импульсного клапана.Импульсный клапан затем закрывался через 5 с, и базовое давление прибора возвращалось к ~ 8 × 10 90 · 107 -9 90 · 108 торр через 95 с. Снижение заряда посредством переноса протона осуществлялось путем улавливания гомодимеров цитохрома c с зарядовыми состояниями D19 – D17 в приборе до тех пор, пока столкновения с остаточными молекулами триэтиламина не снижали зарядовое состояние димеров в течение 100 с, так что 13+ было самая распространенная форма димера. Вновь образованные ионы D13 были изолированы с использованием коррелированных свип-сигналов и диссоциированы с помощью длительной диссоциации, активируемой столкновениями (SORI-CAD) [58] вне резонанса и вне резонанса, при +600 Гц вне резонанса и 3.5 В пик – пик в течение 0,25 с после подачи импульса азота, повышающего давление в вакуумной камере до ~ 10 –6 торр.
Газофазный H / D обмен белковых комплексов
Газофазный H / D обмен гомодимеров цитохрома c осуществляли путем улавливания всех генерируемых электрораспылением ионов в ячейке и введения ND 3 в вакуумную камеру с использованием ручной герметичный клапан (Varian, Lexington, MA) на 180 с при нескольких давлениях в диапазоне от ~ 1 × 10 −8 до максимального ~ 3 × 10 −7 торр.После 180 с замены клапан утечки был закрыт, и вакуумная камера откачивалась в течение 60–120 с, пока базовое давление не стало достаточно низким для изотопного разрешения (<1 × 10 –8 торр). Предыдущие эксперименты по газофазному обмену H / D с белками использовали D 2 O в качестве обменного реагента [22–24, 59]. Из-за трудностей, связанных с длительным временем улавливания (> 300 с) и значительным временем, необходимым для удаления относительно нелетучего D 2 O, в качестве обменного реагента был выбран ND 3 .Давление вводимого в вакуумную камеру НД 3 измерялось некалиброванным ионным манометром, расположенным на расстоянии ~ 1,1 м от ионной ячейки.
Результаты и обсуждение
Диссоциация смешанных
димеров α -лактальбуминаИспользуя растворы ESI с высокими концентрациями белка, можно образовать как гомодимеры окисленного белка α -лактальбумина (каждый белок имеет четыре интактных дисульфидных связи), так и окисленно-восстановленные гетеродимеры α -лактальбумина (один белок имеет четыре интактных дисульфидных связи, а другой – четыре восстановленные дисульфидные связи, причем все восемь остатков цистеина ацетамидированы).И окисленные гомодимеры, и окисленно-восстановленные гетеродимеры образуются непосредственно из ESI с 15 присоединенными протонами ((Ox-Ox) 15 и (Ox-Red) 15, соответственно) (). (Ox-Ox) 15 диссоциирует с довольно широким, но симметричным распределением заряда (), аналогичным тому, что наблюдалось ранее для (Ox-Ox) 13 и (Red-Red) 17 [56]. (Red-Red) 13, как было ранее показано, асимметрично диссоциирует с преимущественным образованием M9 / M4 [56]. В отличие от (Ox-Ox) 15, (Ox-Red) 15 диссоциирует с сильно асимметричным распределением заряда ().Наиболее распространенным процессом диссоциации димера является (Ox-Red) 15 → Red11 + Ox4, процесс разделения, при котором восстановленный α -лактальбумин удаляет почти всего заряда! Обратите внимание, что большая интенсивность сигнала Red11 по сравнению с Ox4 обусловлена как более низким уровнем заряда последнего (сигнал FT-ICR пропорционален заряду), так и несколько меньшей эффективностью обнаружения при более высоком m / z .
( a ) Масс-спектр ESI, полученный из 200 мкМ раствора M (общая концентрация белка), содержащего окисленный (нетронутый дисульфид) и восстановленный α -лактальбумин (1: 1 вода: метанол + 2% уксусная кислота) , ( b ) МС / МС спектр окисленного-восстановленного гетеродимера α -лактальбумина с 15 протонами, (Ox-Red) 15 и ( c ) МС / МС спектр окисленного α -лактальбумина -окисленный гомодимер (Ox-Ox) 15, образованный из раствора ~ 125 мкМ М окисленного α -лактальбумина в 1: 1 вода: метанол + 2% уксусная кислота.Red8 представляет (восстановленный α -лактальбумин + 8H) 8+ и т. Д.
Происхождение сильно асимметричного распределения заряда может быть связано с асимметричным разделением, которое происходит в газовой фазе [56], или с асимметрией конформации или заряд мономеров, составляющих димер, либо в растворе, либо индуцированный процессами электрораспыления. В первом механизме асимметричное распределение заряда происходит из-за конформационного изменения одной из субъединиц мономера, которая составляет димер, после активации.Когда мономер разворачивается, молекула преодолевает активационный барьер для разворачивания и разделения. Из-за кулоновской энергии выход из этого барьера является сильно отталкивающим, и когда одна часть разворачивающейся молекулы выходит из этого канала, она будет управлять разворачиванием остальной части молекулы, когда она отделяется от другой субъединицы мономера. В этом процессе протоны будут переходить к выходящему разворачивающемуся иону, чтобы минимизировать кулоновскую энергию [30]. Поскольку восстановленный белок более гибок конформационно и может разворачиваться в значительно большей степени, чем окисленный белок, восстановленный белок разворачивается и уносит больший заряд, чем окисленная форма.
Альтернативный механизм этой асимметрии заряда заключается в том, что (Ox-Red) 15 может образовываться в растворе путем агрегации сильно заряженных восстановленных и низкозарядных окисленных мономеров α -лактальбумина в растворе или образовываться во время электрораспыления за счет асимметричной зарядки. отдельных белковых составляющих на заключительных этапах ESI [60]. Также было высказано предположение, что асимметричное распределение заряда для гетеродимеров может быть связано с различиями в pI [61, 62]. Хотя возникает разница в зарядке этих двух молекул, она не учитывает величину асимметричного разделения заряда.В масс-спектре ESI смешанного раствора α -лактальбумина восстановленный α -лактальбумин появляется преимущественно при 8+ и в меньшей степени при 9+ и 10+ (). Наблюдается очень мало 11+. Окисленный α -лактальбумин встречается только в зарядовом состоянии 8+. В спектре диссоциации (Ox-Red) 15 доминирующими наблюдаемыми видами являются Red11 и Ox5, несмотря на то, что численность этих видов, продуцируемых непосредственно ESI, незначительна. Хотя можно было бы рационализировать более низкое зарядовое состояние, наблюдаемое для Ox, как немного более низкое зарядовое состояние димера по сравнению с суммой зарядовых состояний отдельных частиц, это не может быть верным для восстановленных частиц, которые имеют значительно более высокий заряд при диссоциации. спектра, чем при формировании непосредственно электрораспылением.Таким образом, этот результат трудно рационализировать, основываясь только на модели, в которой асимметричное распределение заряда является либо реликтом агрегации фазы раствора, либо артефактом процесса электрораспыления. Результаты гетеродимеров (Ox-Red) 15 α -лактальбумина, по-видимому, лучше всего подтверждают процесс асимметричного распределения заряда в газовой фазе [56].
Диссоциация димеров цитохрома c
Гомодимеры цитохрома c легко образуются из растворов для электрораспыления высокой концентрации (~ 100 мкм M) с зарядовыми состояниями в диапазоне от [(цитохром c ) 2 + 19H] 19 + к [(цитохром c ) 2 + 13H] 13+ (сокращенно D19 – D13).Как было показано ранее, диссоциация цитохрома c D19, образованного непосредственно из раствора, приводит к узкому симметричному процессу разделения заряда [56]. МС / МС D17, образованного непосредственно из раствора, приводит к относительно широкому, но симметричному процессу разделения заряда (преимущественно D17 → M9 + M8) ().
МС / МС спектры ( a ) (димеры цитохрома c + 17H) 17+ , (D17), образованные непосредственно ESI, ( b ) (димеры цитохрома c + 13H) 13+ , (D13), образованный выделением D19-D17, восстановлением газофазного переноса протона диэтиламином, выделением и последующей активацией D13, и ионы ( c ) D13, образованные непосредственно из раствора.Ионы образовывались из 100 мМ раствора (вода: метанол, 1: 1 + 2% уксусная кислота).
Напротив, диссоциация D13, образованного непосредственно из раствора, приводит к сильно асимметричному распределению заряда с центром вокруг D13 → M9 + M4 (). Ионы D17-D19 были изолированы и прореагировали с триэтиламином внутри ионной ячейки FT-ICR в течение 100 с, что привело к образованию преимущественно ионов D13. Выделение D13 и последующая активация этих ионов дает спектр МС / МС, показанный на. Доминирующим процессом является симметричная диссоциация с центром D13 → M7 + M6.Таким образом, процесс диссоциации D13 явно зависит от того, образуются ли они в результате восстановления с переносом протона в газовой фазе D17 – D19 () или образуются непосредственно из раствора (). В этих экспериментальных условиях внутренняя энергия иона уравновешивается с полем излучения черного тела при комнатной температуре до активации иона [63]. Таким образом, различие во фрагментации не может быть артефактом различия внутренней энергии. Эти результаты показывают, что структуры ионов D13 различаются для этих двух методов образования ионов и что структура димера влияет на то, как эти димеры диссоциируют, т.е.е., через симметричное и асимметричное разделение заряда. Следует отметить, что димеризация в растворе неспецифична и что может присутствовать большой ансамбль димерных структур. Однако ясно, что существуют по крайней мере две отдельные структуры или различные ансамбли структур.
«Память» ионов D13 может быть связана с различными димерными структурами, существующими в растворе. Эти димеры могут получать разное количество зарядов в процессе ионизации электрораспылением, или процесс электрораспыления может вызывать конформационные изменения в некоторой части димеров при переходе из раствора в газовую фазу, что приводит к различным структурам димеров для D17 по сравнению с D13.Мы не можем четко различать эти две возможности, основываясь только на этих результатах. Однако широкое распределение в зарядовых состояниях димеров, образованных в этих денатурирующих условиях, по сравнению с типичным единственным доминирующим зарядовым состоянием димера, наблюдаемым при электрораспылении из растворов, в которых эти молекулы имеют нативную структуру, предполагает, что первый механизм более вероятен, т. Е. в растворе существуют по крайней мере две отдельные структуры или ансамбль структур, и эти структуры в конечном итоге имеют разную степень заряда в процессе электрораспыления.
Аналогичные результаты наблюдаются для ионов D15, образованных из раствора, по сравнению с ионами, образованными переносом протона. Те, которые образуются непосредственно из раствора, демонстрируют бимодальную диссоциацию: симметричный процесс, сосредоточенный вокруг M8 / M7, и асимметричный процесс, сосредоточенный вокруг M10 / M5 (). Восстановление заряда D19 – D17 с образованием более низких зарядовых состояний с последующим выделением и диссоциацией D15 приводит только к симметричной диссоциации с образованием M8 / M7 (). Опять же, эти ионы D15 явно сохраняют «память» о том, образовались ли они в газовой фазе из более высоких зарядовых состояний, которые симметрично диссоциируют, или непосредственно из раствора.
МС / МС спектры (цитохрома c димеры + 15H) 15+ , (D15), образованного ( a ) непосредственно ESI и ( b ), образованного выделением D19-D17, газ- восстановление фазового переноса протона диэтиламином, выделение и последующая активация D15. Ионы образовывались из 100 мМ раствора (вода: метанол, 1: 1 + 2% уксусная кислота).
Газофазный H / D обмен мономеров и димеров цитохрома c с ND
3Предыдущие исследования показали, что степень асимметрии заряда цитохрома c D11, образованного непосредственно наноэлектрораспылением с использованием водных растворов, содержащих 100 мМ ацетата аммония нейтральный pH зависел от внутренней энергии, которая вкладывалась в ионы [56].Чтобы попытаться установить, в какой степени различные конформеры могут вносить вклад в наблюдаемую энергетическую зависимость, на D11 были проведены исследования обмена H / D. Тщательная настройка параметров источника ESI, в частности увеличение напряжения смещения на внешнем гексаполе накопления и увеличение времени нагрузки гексаполя, способствует образованию цитохрома c D11 ().
ESI масс-спектр 500 мкМ M конский цитохром c в водном растворе со 100 мМ ацетатом аммония, pH ~ 7, со спектрометром, настроенным на получение максимального количества димеров с зарядами 11+ (D11).
Для этих экспериментов по газофазному H / D обмену давление NH 3 изменялось от 1 × 10 –8 до 3 × 10 –7 торр с постоянным временем реакции 180 с. Для эксперимента при самом высоком давлении реагента (наибольшая степень обмена H / D) () среднее количество атомов водорода, обмениваемых на дейтерий, составляет ~ 170 после вычитания обмена 11 протонов. Ширина распределения составляет примерно 110 Да. Отношение сигнал / шум недостаточно, чтобы приписать вариации интенсивностей изотопов разрешимым конформерам.Напротив, M7 в тех же условиях показывает средний обмен ~ 84 атомов водорода на дейтерий с шириной изотопического распределения ~ 50. Таким образом, как средняя степень обмена, так и ширина распределения димера примерно вдвое больше, чем у мономера. Если бы присутствовала только одна структура, нельзя было бы ожидать, что суммарная максимальная степень обмена и ширина распределения будут линейно увеличиваться с массой иона, если предположить, что степень обмена H / D связана с открытой поверхностью кластера.Эти результаты предполагают присутствие множественных конформеров димеров, которые не разрешаются в этих экспериментах по обмену H / D, что согласуется с нашим ожиданием, что эти ионы D11 образуются в результате неспецифической димеризации молекул циктохрома c в растворе.
Частичные масс-спектры ESI цитохрома c (показана область D11) при тех же условиях раствора, что и раньше, и после газофазного водородно-дейтериевого обмена с ND 3 в течение 180 с при ( b ) 1 × 10 −7 торр и ( c ) 3 × 10 −7 торр.
Обширные исследования газофазного H / D обмена мономеров цитохрома c были проведены McLafferty и соавторами [22–24], которые обнаружили целых семь конформаций цитохрома c M7, образованных непосредственно ESI [24]. В наших экспериментах в качестве обменного реагента используется ND 3 вместо D 2 O [24], и время реакции короче. Распределение изотопов для M7 предполагает наличие нескольких конформеров, согласующихся с результатами, полученными Маклафферти и соавторами.
Выводы
Асимметричное распределение заряда, которое наблюдается при диссоциации гомодимеров белков, можно объяснить двумя различными механизмами. В одном механизме асимметрия индуцируется разворачиванием одного из белков в димере при активации. В димерах, состоящих из двух почти идентичных белков, белок с более высокой конформационной гибкостью отделяется от комплекса с большим зарядом. Это может быть связано с тем, что часть молекулы покидает барьер диссоциации, где дальнейшее развертывание молекулы индуцируется отталкивающим кулоновским потенциалом.Во время этого процесса разворачивания и разделения протоны передаются разворачивающемуся белку, чтобы уменьшить общую кулоновскую энергию. Этот механизм лучше всего объясняет результаты, наблюдаемые для гетеродимеров α -лактабумина, состоящих из одного окисленного и одного восстановленного белка. Этот механизм также лучше всего объясняет результаты для димеров цитохрома c с 11 зарядами, для которых наблюдается энергетическая зависимость в степени асимметрии [56].
Некоторая часть наблюдаемого асимметричного распределения заряда также может быть приписана другому механизму, в котором структуры мономеров в димере различаются.Это лучше всего объясняет результаты, наблюдаемые для димеров цитохрома c с зарядами 17+ по сравнению с димерами с 13+ зарядами. При образовании непосредственно из раствора первый диссоциирует с симметричным распределением заряда, тогда как последний диссоциирует с сильно асимметричным распределением заряда. Однако, когда зарядовое состояние ионов димера 17+ снижается до 13+ в газовой фазе посредством мягких реакций переноса протона, образующиеся ионы 13+ диссоциируют с симметричным распределением заряда.Это ясно демонстрирует, что ионы 13+, образованные из ионов 17+, имеют другую структуру (или разные ансамбли структур), чем те, которые образуются непосредственно из раствора, и что это структурное различие отвечает за асимметричное и симметричное распределение заряда, наблюдаемое при диссоциации этих 13+. ионы. Это первое зарегистрированное свидетельство того, что асимметричное распределение заряда может быть вызвано структурными различиями в субъединицах мономера в комплексе.
Наконец, H / D обмен зарядового состояния 11+ димеров цитохрома c указывает на присутствие множественных конформеров, хотя эти конформеры не разрешаются в этом эксперименте.Дальнейшая работа с другими реагентами для обмена H / D или в других условиях, или с конформационно-селективными методами, такими как подвижность ионов, в сочетании с экспериментами по диссоциации может предоставить дополнительную полезную информацию о процессе разделения заряда. Лучшее понимание этого процесса в конечном итоге улучшит структурную информацию, которая может быть получена при газофазной диссоциации нековалентных комплексов.
Благодарности
Авторы благодарят доктора Дэвида Кинга (Калифорнийский университет, Беркли) за восстановление и очистку α -лактальбумина.Эта работа была поддержана Национальным институтом здравоохранения (грант № R01-GM64712-01).
